
The views expressed in this paper are those of the writer(s) and are not necessarily those of the ARJ Editor or Answers in Genesis.
Introduction
I used to think that when creationists talk about the discussion between creationism and evolution as a clash between two worldviews, they were wrong. Jeanson has helped me change my mind. It is a clash between worldviews: the scientific and the religious. To make it short: in science, no text is infallible. Everything has to be tested against observation. In (some versions of) religion, there is an infallible text (in Christian creationism, of course it is the Bible). If an observation contradicts the text, the observation is by definition wrong. This simple fact leaves creationism as unscientific!
A little food for thought: Jeanson’s response is about four times the length of my review! You could say that refusing nonsense with truth is more time-consuming than stating nonsense. Perhaps refusing truth with nonsense is even more time-consuming. It should be easier to argue in favor of truth than to have to make up flawed arguments in favor of nonsense!
I urge any reader, creationist or otherwise, to contact me if they need clarification of one or more points in this, rather short, response.
Any clarification of genetic terms or principles can be studied in Jeanson’s book, which has a brilliant account of genetics.
I have to admit that I should have been more systematic in my review. Too often, I do not explicitly mention what chapter I am talking about. This causes some confusion.
I have tried to keep my reply short—not that I have succeeded. Instead of taking Jeanson’s objection point by point, I’ll make some general comments on why I do not think Jeanson has much new to offer. Some points, though, I feel need more thorough comments.
To fully appreciate this reply, first read my review (Frello 2018) and Jeanson’s response (Jeanson 2018).
Here is an introduction to a guiding principle in science, which is useful to know, and which the reader is invited to use whenever it seems appropriate: Occam’s razor: a principle stating that when choosing among alternative theories, we should prefer the one with fewest arbitrary assumptions. Of course, we should accept assumptions that seem well supported. In genetics, one such extremely well supported assumption is the theory of the transcription-translation system from DNA to protein. Occam’s razor does not state that there always will be one, and only one, such theory. It might depend on what you accept as well supported assumptions.
On “Introduction” and “Overview”
Jeanson refers to our discussion about the reliability of ancient mtDNA (Frello 2017a, b; Jeanson 2017a ,b). I urge our readers to read the articles, and judge for themselves if I have “revealed the deficiency of [my] best anti-YEC claims” as Jeanson will have it. In Frello 2017a and Jeanson 2017a special attention should be paid to the terms “contamination” and “degradation.” In Jeanson 2017b, special attention should be paid to considering whether Jeanson actually argues against my suggestion in Frello 2017b (to confront the experts within the field of ancient human DNA).
On “Frello’s General Claims”
In short Jeanson summarizes the three parts of his book as follows:
- The question of origin of species is fundamentally a genetic question. That’s why genetics is such an important tool in the study of evolution. I fully agree, which should be clear from my review. Not knowing genetics, Darwin took a massive scientific risk, when he published On the Origin of Species. I do not argue against that.
- Darwin’s 1859 data are mostly irrelevant today. So what!
YEC endorses migration as an explanation for biogeography. I comment on that.
YEC endorses speciation. I comment on that.
YEC’s explanation for the pattern of groupings of life have matured. I comment on that. - YEC outstrips evolution in genetics. I comment on that.
The rest of point 3 is a clarification of this statement.
Why Jeanson calls these comments (the vast majority of the review) a side step from direct confrontation of the main claims of his book, is beyond me.
On “Frello’s Claims About Biogeography”
Here Jeanson goes to some length in explaining how the situation was in 1859. Except that Darwin used biogeography as one of his arguments in favor of evolution, the situation back then is not very important. For example, Jeanson repeatedly mentions species fixity as one of the ideas creationists have given up. In my review, I do not mention fixity at all. Who cares about outdated ideas?
Jeanson complains about my negligence in not reading the references found in an Endnote to Chapter 4. Sorry Dr. Jeanson. If you have an important argument, do not put it in an Endnote, and especially not in references to which you only give a useless four-line review. That kind of trap is telling about Jeanson’s strategy. What Jeanson is actually asking his reader is to read 400 endnotes and look up and read hundreds of papers, webpages, and other references to see if some important clue was hidden somewhere. Hardly the strategy of a person who honestly wants to inform his reader.
I think Jeanson’s statement “neither the creationist position nor the evolutionary model has a consistent, comprehensive, discipline-wide explanation for biogeography” is fair. Nothing in my review talks against this view. From Jeanson’s YEC point of view, it is a “historical contingency” that of 19 families of marsupials, 17 are endemic to Australia and the nearby islands! I call it a “coincidence” to Jeanson’s discomfort.
I point to two more striking facts: Four different families of Monkeys (the group Platyrrhini) ended up in South America. Four different families of Lemurs (the group Lemuriformes) all ended up on Madagascar! In Chapter 10, Jeanson equates family with biblical kind, but here Jeanson’s answer is that a family is not necessarily equal to kind. The identification of kinds is still a [guess]work in progress. More on that in the section Speciation.
I do not conclude, as Jeanson will have it, that evolution at present can explain biogeography in all its details. I conclude that “Jeanson fails to account for biogeography. . . .”
In spite of all Jeanson’s words, his position still necessarily is that Biogeography can be explained by migration out of Eurasia (Mt. Ararat), and mine that this is an unfounded position.
On “Frello’s Claims About Taxonomy”
Jeanson thinks I misrepresent his position, “. . . that both evolution and creationism predict hierarchies.” But how is that any different from my reference to Jeanson’s position being that: “. . .common descent is not needed to explain the nested hierarchies”?
Jeanson doubts that I will reject an often mentioned argument for evolution: The universal genetic code.1 Well Dr. Jeanson, I have news for you: I do reject it! That’s why I didn’t mention it in my review. Now that Jeanson has opened this discussion, let’s see where it leads. The common genetic code (the nuclear code) is an equally good argument for common design as for common ancestry, and therefore an argument for neither. It is in fact the mitochondrial genetic code, which can be used as an argument for common ancestry. Not because they are identical, but because they are different. Mammals have one code, Insects a slightly different one, Fungi yet another. More than ten slightly different codes are known at present. Why would a designer use different codes in different organisms, and why would the differences follow groups of organisms, otherwise accepted to be closely related? From an evolutionary point of view, this is easy to understand. The mitochondrial genome (mtDNA) has only very few protein coding genes (13 in most animals). Therefore a code-changing mutation has a much better chance of not being lethal here than in most other genomes. A code- changing mutation in the nuclear genome (with tens of thousands of genes) would be lethal, because it would change the amino acid sequence of so many vital genes that at least some are bound to have their function destroyed.
In my view, our fundamental disagreement is the following: What does it take for a taxonomy to be more than an arbitrary personal opinion.
Evolution suggests one, and only one, foundation for taxonomy: Common descent. YEC (or more precisely: the idea that living organisms are designed, and groups of organisms above the level of “created kinds” therefore are genetically unrelated) cannot suggest any such unique foundation for taxonomy. Jeanson tries to do so for designed objects, vehicles. He suggests that vehicles should be placed in two large groups: powered vs. non-powered. But he cannot, and does not, offer any explanation to why this should be a better criterion than any other. In Frello 2018, I mention military vs. civilian; for transportation of persons vs. for transportation of goods as examples of alternative criterion. Another suggestion could be by brand. Why even group vehicles together? Why not all powered, designed objects vs. non-powered objects? Anything goes. None are natural, all are cultural.
Common descent immediately suggests that we should look for a nested hierarchy of groups-within- groups of organisms. Talking about multicellular organisms, there can be only one such correct hierarchy: The one that reflects common descent (at least above the genus level, where hybridization becomes very implausible).
Even if we accept Jeanson’s arbitrary suggestion of powered vs. non-powered vehicles; we still do not have a unique system beneath this level. If powered defines a group, it seems reasonable that the type of engine should define the next, lower, level. But Jeanson suggests land vs. air vs. sea instead. This choice again is completely arbitrary.
In biology, as a consequence of common descent, the science of taxonomy therefore becomes the science of identification of the nested hierarchy of groups of organism. From this, it follows that one kind of information beats all others, when it comes to identification of such groups: DNA. It is easy to see why: groups are defined by common ancestry. Ancestry is equal to genetic ancestry. Genetic information is stored in DNA.
In YEC, taxonomy becomes the arbitrary choice of groups. Arbitrary at all levels. Based on an equally arbitrary choice of traits. If this is what Jeanson thinks qualifies as a scientific argument in favor of a taxonomy for designed objects, it is no wonder that creationism is completely ignored by mainstream scientists as irrelevant.
To point it out more unambiguously: whenever possible, DNA should be (and is) used for identification of groups. Not fur-color or -structure, not reproductive organs, not general appearance, or any other physiological or anatomical trait. Dealing with groups where DNA is not available (especially fossils), physical traits have to be used. Again, a guiding principle can be found: traits that are difficult to change without disrupting survival or reproduction, should be preferred. Jeanson goes to some length ridiculing the identification of such traits, all in vain.
Jeanson thinks I concede that not all genes suggest the same phylogeny. I simply state a fact. As Jeanson knows, contradicting phylogenies are mostly found between closely related species, and can be understood as “incomplete lineage sorting” or as the result of the stochastic nature of mutations. Using large groups of genes solves this problem.
All in all, if all living organisms evolved from a common ancestor, we should expect to be able to group living organisms according to one natural criteria: common ancestry, based on genetics as the most reliable source of information. If living organisms were designed, no such natural criterion or basic source of information should be expected to be found. Judge for yourself.
Jeanson thinks that the reason I do not comment on transitional forms, homologous structures, or vestigial structures is that I agree with his arguments. Let me immediately free him from his delusion.
Regarding transitional forms. Why would a designer construct several transitional forms between a land animal and a whale (e.g. in terms of hind legs and the position of nostrils), just to see them go extinct within a few thousand years from their creation, and for no obvious reason? I guess I need not explain why transitional forms are expected, if we accept evolution.
Regarding homologous structures. I do not recall reading about that in the book. The search engine (I have the Kindle-version of the book) doesn’t find the term. Perhaps Jeanson talks about it under a different term.
Regarding vestigial structures. First we have to agree upon what that means. According to NatureEducation,2 vestigial describes “. . . something occurring in a simpler, less functional state; sometimes a remnant of a larger more robust form.” It clearly does not mean “purposeless leftovers of evolution” as Jeanson has it in his book. This nullifies Jenson’s arguments.
On “Frello’s Claims About Genetic Diversity”
I have to admit that Jeanson is right in his criticism that my treatment of this topic is less than rigorous, and that I tend to confuse the information given in Chapters 7–10. So let me try to clear out the points on which Jeanson thinks I am ambiguous or misrepresenting him.
First, let me clarify my use of the term homology. As Jeanson assumes I mean “percent relative genetic identity.” The alternative being absolute instead of relative.
One of Jeanson’s conclusions in Chapter 8 is that most variations in nuclear DNA, found in organisms that share a common ancestor, are inherited from variation in that common ancestor. The logic of the analyses in Chapter 10 is that all the variation in mtDNA, found in organisms that share a common ancestor, are due to mutations. I agree on both points. That is an uncontroversial position from an evolutionary point of view.
mtDNA tells an unambiguous story. As stated above, Jeanson accepts that homology in mtDNA can be used as a measure of the distance to a common ancestor. If mtDNA from Humans, Chimps, Gorillas, and Orangutans are compared, the pattern is clear.
Jeanson accepts the relationship between Chimpanzees, Gorillas, and Orangutans, identifying them as members of the family Pongidae, (which is no longer accepted in mainstream taxonomy). But from a genetic point of view, it is unfounded to accept the relationship between these three genera, leaving out humans. The only reason he does so, is the YEC assumption that the Bible is infallible (as mentioned, scientists would never accept any text as infallible). As indicated in fig. 1, mtDNA strongly suggests that Chimpanzees are closer related to Humans than to Gorillas or Orangutans.
Jeanson accepts that genetic homology can be used to indicate relationship within kinds, but apparently not in this case!
Actually there is no clear demarcation between within kind and between kinds when it comes to genetic homology. I’ll return to this in the section on speciation.
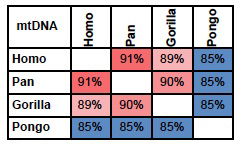
Fig. 1. Percent identity between mtDNA from Humans
(Homo); Chimpanzees and Bonobos (Pan); Western and
Eastern Gorillas (Gorilla) and Bornean and Sumatran
Orangutans (Pongo). Colors indicate level of homology:
Red: high; Blue: low.
Jeanson has some partly relevant comments on fig. 1 in Frello 2018. (Mutations accumulated since the Flood in various species of the Cat-family, Felidae, Johnson et al. 2006.) To explain this I first have to explain a little genetics. According to NatureEducation,3 the term allele refers to an alternative form of a gene not alternative single nucleotides, as Jeanson will have it. For the sake of clarity, let’s call alternative forms of a gene, gene-alleles, and alternative single nucleotides, nt-alleles. Each individual has two copies of each gene. From a YEC point of view, all Cats descend from two individuals on board the Ark. Together these two individuals therefore had a maximum of four different copies, gene-alleles, of each gene. The problem is then to identify groups of species with genes, which descend from the same gene-allele. Differences within such gene-alleles must be due to mutations that have occurred since the Flood.
Jeanson’s point is now that there can be a multitude of differences in the DNA-sequence of two gene-alleles. This is absolutely correct, and I have not argued against it. What I have done in fig. 1 in Frello 2018 is to add the differences between species of Cats for 15 different genes, and identify four groups of most identical species; each supposedly representing the descendants of one original gene-allele per gene. By doing so, I risk mixing a number of differences within gene-alleles (which is relevant), with a number of differences between gene-alleles (which is not relevant). It is not clear from Jeanson 2018 that this is what he thinks is wrong with my calculations, but it is the only way I can make sense of his objections. Jeanson is correct that this gives a wrong picture, and I have therefore attacked the problem in another, more correct, way, looking at single genes. Fig. 2 shows the variable positions in the gene SIL. The results clearly show that a considerable number of mutations is necessary to explain the pattern of sequence variation. A total of 22 mutations is found (not taking selection into consideration). The largest number of mutations in a single group is 11. If the mutation rate in Cats is comparable to that of Humans, the probability of finding this number of mutations in this dataset is negligible.4 Therefore, the conclusion is still correct: Jeanson’s suggestion that present day variation among species is due to the distribution of different original gene-alleles, cannot explain the variation among modern species.
Of course, a single example is not proof, though in this case it is a very strong indication. I therefore invite Jeanson (or any of our readers) to repeat the analyses for the rest of the genes in Johnson et al. 20065 or in any other nuclear gene, sequenced in several closely related species. (Kinds that, according to YEC, were either not in the Ark (e.g. aquatic Mammals) or were present in more than one couple (e.g. Bovidae) cannot be used in such analyses).
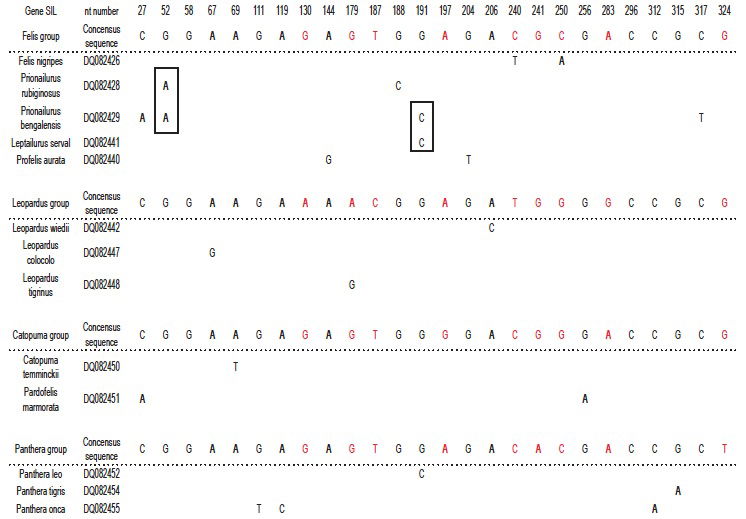
Fig. 2. YEC-interpretation of 12 copies of a partial sequence of the SIL-gene from species in the Cat-family, Felidae. Only variable nucleotides are shown. The four gene-alleles are indicated as the four groups (identified to be the combination of sequences that result in lowest number of deviations from the consensus of each gene-allele). In each group, a consensus sequence is identified as the one that results in the lowest assumed number of mutations. Differences between the four consensus sequences are indicated in red. Only those nucleotides that deviate from the relevant consensus sequence are indicated. Deviations that might originate from a single mutation are in boxes. Only species with unique deviations from the relevant consensus sequence are shown (12 out of an original set of 38 species). To explain the results the following minimum numbers of mutations are necessary: Felis group: 11. Leopardus group: 3. Catopuma group: 3. Panthera group: 5.
On “Multifunctionality of Mitochondrial Genes”
Just to make sure that our reader knows what we are talking about, most animal mitochondrial genomes (mtDNAs) contain 13 protein-coding genes, 2 rRNA-coding genes, 20+ tRNA coding-genes, and the so-called control region or D-Loop. The mitochondrial protein coding genes deal with some of the most fundamental biochemistry in the organisms. Genes with the same biochemical functions (and most often with recognizable amino acid sequence) are found throughout not only the animal kingdom, but in plants, fungi, and the plethora of unicellular Eukaryotes. Even in bacteria.
Jeanson quotes his 2013 article, where he accepts that the hierarchy suggested by mtDNA homology reflects the one suggested by the Linnaean classification system. He then states that this system is based on function. I assume he is mainly talking about anatomical and physiological function, as Linné hardly knew any biochemistry. His conclusion is to ascribe such function to the genes of mtDNA. Just to clarify, evolutionary theory offers a much simpler explanation: homology is due to common descent at all levels.
Jeanson then argues that he does not necessarily suggest multiple anatomic/physiological functions of each gene. Instead, optimization6 of the known function in the various organismal contexts could be relevant.7 I agree that optimization is a very likely scenario. However, he makes no argument, as to why such optimization should reflect the same hierarchies, regardless of what gene you are looking at. And why should it be the same hierarchy suggested by anatomy/physiology?
Jeanson makes no attempt, neither in this response, nor anywhere else to my knowledge, to explain why mtDNA from Horses, Tapirs, and Rhinos (order: Perissodactyla) should be more identical to each other, than mtDNA from, say, Horse and Cow, or Rhino and Elephant, if the only function refers to optimization. What kind of relevant optimization could result in this striking pattern?
He then goes on to the kind of function I was referring to—anatomically and physiologically relevant function. To my great (admitted, malicious) pleasure, he cites his own suggestion (Jeanson 2013), that the ATP6-gene could have some function in egg laying. This is, pardon my French—nonsense! The ATP6-gene clusters bony fish, amphibians, birds, and Reptilia. But some mammals, Monotremes, lay eggs; and some fishes, Cyprinodonts, give birth to live young. If Jeanson’s idea was right, we should expect Monotremes to group with fish and the rest, and Cyprinodonts to group with Mammals (or, alternatively, form separate groups). They do not. They end up in the system exactly where they should according to evolution. Trust me, I have checked. Or check for yourself.8
I have no intention to reject that proteins can have multiple functions. I was specifically talking about the mitochondrial genes. Here Occam’s razor is relevant again.
Jeanson’s hypothesis: protein coding genes in the mtDNA have more than one function!
Alternative hypothesis: protein coding genes in the mtDNA have only one function—the known one. The burden of proof is clearly on Jeanson to show that such multiple functions exist, not on me to show that they don’t.
Jeanson quotes my example, using the Cox1 gene, describing the difficulties multifunctionality faces. He then paraphrases the quotation substituting “Jeanson” with “taxonomist” and “Cox1” with “skull shape.” The point being that when I challenge Jeanson to explain the function of Cox1, he could challenge a taxonomist to explain variation in skull shape. Again, Jeanson fails completely.
Not all differences in anatomical details are functional. Some are, some are not, so you cannot always expect such explanations to exist. At the end, Jeanson’s paraphrase turns into pure nonsense, as he has to include Ladybird, Thale cress, Portobello, and the Malarial parasite, because I do. None have a skull. They all have a Cox1 gene, though. My arguments hold; Jeanson’s paraphrase is weak from the start, and becomes nonsense towards the end.
I cannot wait for “the functions for these sequences [to] be discovered” as Jeanson writes.
Next on silent mutations (mutations that change the DNA, but not the protein). I never stated that silent mutations never are functional! I know they are, probably by influencing the level of expression. Jeanson doesn’t answer my most important question, though: why functional codon9-use should be expected to reflect taxonomy.
On “Selection”
Jeanson starts by quoting me: “In all cases [of analysis of mtDNA], [Jeanson] fails to include selection, though this can be shown to be a very real phenomenon.” I should of course have been specific about what I was thinking of. My comment is aimed at Chapter 7, figures 7.3–7.6 and 7.18–7.25, where Jeanson makes a number of calculations of the predicted number of differences between mtDNAs from different specimens of various species, both under YEC and under evolution. In these calculations, Jeanson does not refer to selection.
Of course, Jeanson is right that attempts should be made to make rigorous predictions of the effect of selection. However, to dismiss the potential of selection because its strength is unknown is unsubstantiated. The right conclusion must be that Jeanson’s figures actually do not support either model over the other. Jeanson urges me to look for errors in his calculations. Why should I? Interesting as they are, they cannot be used to distinguish between evolution and YEC.
Turning to the reliability of ancient human DNA, Jeanson rejects my claim that “mistakes [due to degradation] in the sequence should be expected to be randomly distributed, when counted as synonymous10 vs non-synonymous substitutions”11 by suggesting the opposite: that degradation could in some way be directed towards synonymous substitutions.
The answer to this is rather technical, so bear with me. First, we have to appreciate that due to the way proteins are coded by DNA, non-synonymous mutations are much more likely to occur than synonymous. Nevertheless, the vast majority of differences between Neanderthals and modern Humans, are synonymous. This is the phenomenon we aim at explaining.
I suggest that the sequences of Neanderthal mtDNA are reliable, and the surplus of synonymous substitutions is due to strong selection against the non-synonymous. Jeanson suggests that it is due to a non-random distribution of errors in the sequencing, due to degradation.
As an example, one-way degradation could be non-random is if it had a strong tendency to result in A being misread as some other nucleotide. Let’s consider the sequence GCACTATCAATGTA and what happens if it read as GCCCTTTCCGTGTG. What number of synonymous and non-synonymous substitutions would this result in? The answer is that no one can say. It depends entirely on the so-called reading frame. Remember the term codon!12 A reading frame is the way the sequence is split into codons.
In fig. 3 I have translated the original and the degraded sequence into amino acids (three letter code), for the three possible reading frames. For each I have indicated the resulting number of synonymous and non-synonymous changes.
As the vast majority of the DNA sequence of most genes is positioned far (more than 100 nucleotides) from the start-codon that defines the reading frame, it is hard (I would say impossible) to see how a degrading mechanism could be directed towards synonymous or non-synonymous errors. Adding to Jeanson’s difficulties is the fact that the start codon: ATG, which defines the reading frame, easily can occur out of frame in the sequence of the gene, as in the example. If the degradation was somehow directed by this ATG sequence, how could it distinguish between ATG as a start codon or as an out-of-frame sequence within the gene? The translation system is actually a rather sophisticated mechanism that can identify the right ATG start codons.
Again, we can apply Occam’s razor (remember, we aim at explaining the surplus of synonymous substitution).
Jeanson’s hypothesis: A mechanism directs degradation of DNA towards synonymous substitutions.
My hypothesis: there is no such mechanism.
I think it is clear that a very heavy burden of proof lies on Jeanson.
As a last rescue device, Jeanson suggests hypermutation. Sigh!
Again, Occam’s razor would put the burden of proof on Jeanson.
In Frello 2017a, which is all about the reliability of ancient human DNA, I do not mention this argument, and Jeanson thinks it is a change of subject that I mention it in my review. It obviously is not. The subject always was the reliability of ancient human DNA sequences. It is simply a new argument in the same debate, and Jeanson fails to give a relevant reply.
On “Frello’s Claims About Speciation”
First on breeds of domesticated animals vs. speciation in the wild: Jeanson asserts that evolution of species within a kind in a biblical timescale, is unproblematic, because breeds of, say, horses and dogs are more deviating than species of the same family. I tested this idea in the Dog family (Canidae) on the most informative level on information: DNA.

Fig. 3. Translation of three different reading frames of two slightly different DNA sequences. Translation into three-letter amino-acid code is indicated. Number of non-synonymous and synonymous changes are indicated.
In my review (Frello 2018), I find that mtDNA from the Dhole and the African wild dog has 15 times more substitutions than mtDNA from Domesticated dog and Wolf. I conclude that Jeanson’s assertion is unsubstantiated. Jeanson then makes a kind of backwards ad hominem argument: that I should accept this argument because it comes from Darwin himself (his highlighting). Why would I? I don’t regard Darwin as an infallible source of information. I’m not religious!
Jeanson states that because I refuse to accept the breed-species comparison as an argument for short timescales, I should refuse it for common ancestry too. On what bases? Differences in DNA is a good, though far from perfect, indicator of timescales. DNA indicates that the timescale of speciation is orders of magnitude longer than that of breeds within a species.
In the paragraph starting with “Fifth . . .” Jeanson calls it a misrepresentation that I state that he doesn’t make any calculations to support his claims. Reading my review, there can be no doubt this remark is directly aimed at the breed-species example I just went through. And there, it is relevant, whether Jeanson likes or not. In spite of all his words, Jeanson fails to refute the conclusion from the Dog family example.
In the quotation on the right column of page 80, I have to admit to a blunder. I make a remark that Jeanson doesn’t include extinction, though he accepts it as a very real phenomenon. Unfortunately, I have mixed the results from two chapters. Chapter 7 is on accumulation of mutations, Chapter 10 is on linear rates of speciation.
The calculations of mutations since last common ancestor (Chapter 7) is where my remarks on extinction and distance to last common ancestor is relevant. Jeanson uses extant data on mutation rates to calculate the number of mutations that should have occurred since the first appearance of the genus/species in the fossil record. As I have pointed out above, selection makes such calculations rather useless, but what I point out here, is that the first appearance in the fossil record might not be the last common ancestor of all extant species. This is still a relevant objection.
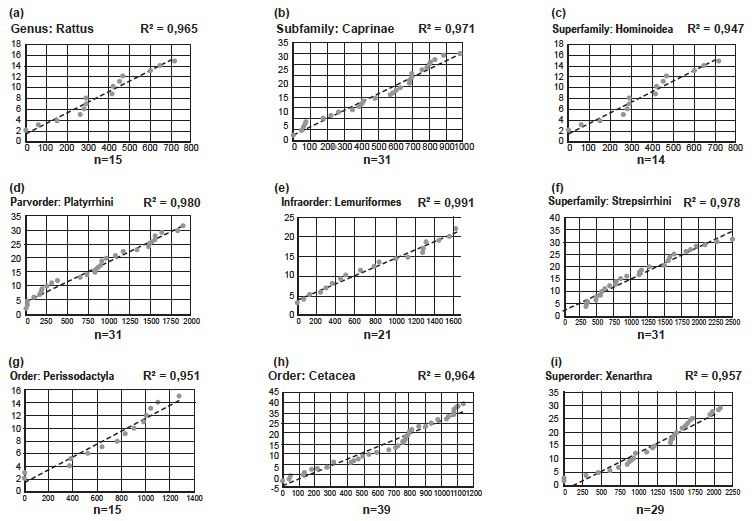
Fig. 4. Results of analyses of speciation in nine groups13 of mammals, all showing a linear correlation between number of substitutions and number of species. n indicates the number of species in each analysis. R2-values are indicated in each graph. (a) Genus: Rattus. (b) Subfamily: Caprinae. (c) Superfamily: Hominoidea. (d) Parvorder: Platyrrhini. (e) Infraorder: Lemuriformes. (f) Suborder: Strepsirrhini. (g) Order: Perissodactyla. (h) Order: Cetacea. (i) Superorder: Xenarthra.
Back to Chapter 10. There is a telling sentence about one page into the chapter “. . . creationists and evolutionists disagree on ancestry above the level of family . . . .” This only makes sense if Jeanson identifies the biblical kind as the family-level. This is contrary to his claims in the section on biogeography (Jeanson 2018), where he calls the identification of kinds a work in progress. Also, in his comments on biogeography, he suggests that the four families of Platyrrhini, South American monkeys, might be one kind, and Lemuriformes, the four families of Lemurs, another. This too contradicts families as kinds.
In figs. 10.2 and 10.4–10.24 Jeanson identifies a linear correlation between number of mutations and speciation, within various vertebrate families. As the identification of kinds is a work in progress, we could have expected him, at least just for the sake of the argument, to have included an analysis of Platyrrhini and Lemuriformes (see section Biogeography).
Jeanson reaches a general conclusion: the pattern of speciation within each family can be explained by a linear model, which can be explained from a YEC point of view, if we accept the family level as the biblical kinds.
But is this conclusion sound? Strangely, Jeanson makes no attempt to test patterns of speciation at other taxonomical levels. Strange because if the identification of kinds is a work in progress and the linear pattern of speciation has any relevance to the identification of kinds, it would be urgent to see if the pattern is repeated at other taxonomical levels.
I have made a few similar analyses of my own. For a description of method: See Jeanson 2017c, Chapter 10. In such analyses Jeanson accepts R2-values above 0.9 as an indication of correlation, and groups with 13 or more members as sufficiently large for a relevant statistical analyses. My analyses meet both requirements.
I found the linear pattern repeated in the Genus Rattus (Rats), the Subfamily Caprinae (Sheep and Goats part of the Family Bovidae), the Superfamily Hominoidea (Great Apes [including Humans and Gibbons]), the Parvorder Platyrrhini (four families of South American Monkeys), the Infraorder Lemuriformes (four families of Lemurs), the Suborder Strepsirrhini (Lemuriformes and the Loris), the Order Perissodactyla (Horses, Tapirs, and Rhinos), the Order Cetacea (Whales), and the Superorder Xenarthra (Armadillos, Sloths, and Anteaters).
I also repeated two of Jeanson’s analyses—figs. 10.10 and 10.24. I got very similar results, indicating that the analyses were done in the right way.
These results indicate that the pattern, Jeanson found in families, is repeated at all levels from genus to superorder. Of course, a few examples are not enough to make solid conclusions. I therefore invite Jeanson to prove me wrong, by repeating and publishing this line of investigation at various taxonomical levels, throughout the vertebrates, especially mammals.
If we accept evolution, it is no surprise that the same overall pattern of speciation can be found on various taxonomic levels. Such levels are, after all, more or less arbitrary human constructs. Notice, that I do not postulate that evolution predicts a linear pattern, just that if some pattern is found, we should expect it to be repeated between taxonomic levels.
On “Additional Points”
Darwin didn’t know genetics.
In this section Jeanson states that I object to his claim, that “Darwin took a risk when he penned a strong answer to a deeply genetic question—long before genetic data were available to test it.”
I actually do not. I point to a number of possible situations, where knowledge of genetics potentially could, but in practice doesn’t, refute evolution. It is spelled out quite clearly. I do not “seem to agree” that genetics is important, as Jeanson will have it.
Jeanson wants references to my claim that for evolution to work, variation has to be endless, and new traits have to be able to occur. I think Jeanson misunderstands this. It seems as if he reads it as if I state that this actually is an observed fact. But what I state is that for evolution to work, genetics have to have these qualities. I hope we can agree that this is uncontroversial.
On “Strange Quotes”
First, on the neck of the Giraffe: Jeanson misread my text. What I clearly state is that I doubt that major changes are needed (in the developmental pathway for vertebrae in the neck of the Giraffe— my emphasis). In Jeanson’s head, this amounts to stating that no change is needed at all. Sigh!
Next on the “Could jellyfish become jaguars” quotation: this is not a question of biology in general. It is a specific evolutionary question! To what other field of biology could it refer?
Next on Michael Behe: what Behe has to say to his critics is irrelevant. From the context in Jeanson’s book, it is crystal clear that the relevant topic is whether Behe’s ideas are accepted by mainstream science. They are not!
Next on senses: in short my argument is that natural selection increases survival; how can a change in an eye help survival? Only by giving a more accurate picture of the world. Therefore natural selection can explain that we can trust our senses. Jeanson thinks that this is a circular argument, because the only way I can know that natural selection increases survival is by inspection using my senses. But actually, No! In fact, the argument for natural selection to increase survival is purely logical. (See my account of evolution in the section “Darwin didn’t know genetics” in Frello 2018.)
Actually, it is Jeanson who has the problem. He trusts his senses because he believes he was created by a God, who cannot be deceitful. How does he know, God cannot be deceitful? By reading the Bible, using his eyes. Therefore, he trusts his eyes because he trusts his eyes!
Next on my concluding remark: “Would you trust an Atheist to teach your children about Christianity? If not—don’t trust a creationist to tell them about evolution!” It seems as if Jeanson thinks this implies that a scientist is not entitled to criticize a work on science, just because it is written by a creationist. How he reaches this conclusion is beyond me (of course except if he does not think his book is about science!).
Summary and Conclusion
Despite writing a 17,000+ word defense of his book, Jeanson still fails to argue in favor of YEC and against evolution. How Jeanson can call my lengthy comments on all three central theses in his book (points 1 to 3 in the “General claims” section) a side step, I still don’t understand. By not meeting my critique, Jeanson (inadvertently) strengthens the evidence for evolution. All in all, Jeanson’s book fully deserves the fate it has got in the scientific community—silence. (Except for one retired, largely unknown, molecular biologist and creationism nerd—me.)
If the Jeanson/Frello word ratio will be the same in Jeanson’s next response, we can look forward to a 20,000-word response. I cannot wait.
References
Frello, Stefan. 2017a. “On the Creationist View on mtDNA.” Answers Research Journal 10: 181–182.
Frello, Stefan. 2017b. “Reply to “Response to ‘On the Creationist View on mtDNA’.”.” Answers Research Journal 10: 237.
Frello, Stefan. 2018: “No Replacement of Darwin.” Answers Research Journal 11: 57–62.
Jeanson, Nathaniel T. 2013. “Recent, Functionally Diverse Origin for Mitochondrial Genes from ~2700 Metazoan Species.” Answers Research Journal 6: 467–501.
Jeanson, Nathaniel T. 2017a. “Response to “On the Creationist View on mtDNA”.” Answers Research Journal 10: 183–186.
Jeanson, Nathaniel T. 2017b. “Response to “Reply to ‘Response to ‘On the Creationist View on mtDNA’.”.” Answers Research Journal 10: 239–240.
Jeanson, Nathaniel T. 2017c. Replacing Darwin: The New Origin of Species. Green Forest, Arkansas: Master Books.
Jeanson, Nathaniel T. 2018. “Response to “No Replacement of Darwin: A Review of Replacing Darwin—The New Origin of Species”.” Answers Research Journal 11: 63–83.
Johnson, Warren E., Eduardo Eizirik, Jill Pecon-Slattery, William J. Murphy, Agostinho Antunes, Emma Teeling, and Stephen J. O’Brien. 2006. “The Late Miocene Radiation of Modern Felidae: A Genetic Assessment.” Science 311, no.5757: 73–77.


















