

Research conducted by Answers in Genesis staff scientists or sponsored by Answers in Genesis is funded solely by supporters’ donations.
Abstract
During the last 84 years, determinations of the 238U and 235U decay constants and half-lives have been made using direct counting experiments and geological age comparisons, as well as by critical reviews and reevaluations of those determinations. By 1971 the direct counting experiments had provided 238U and 235U half-life values with small uncertainties which ever since have been the recommended values used in all U-Pb age calculations. All the geological age comparison studies have utilized those recommended values, in spite of the admitted philosophical circularity involved. And the critical reviews and reevaluations have all converged on these same recommended values because of the meticulous care taken in the 1971 direct counting experiments, which then gave those experimental results the dominant weight in the calculation of mean values. But there have still been repeated calls for more modern, more accurate direct counting experiments to more precisely determine the 238U and 235U half-lives. It is difficult to determine precise values for the 238U and 235U half-lives because the 238U and 235U peaks in the a-energy spectrum have to be accurately delineated where they overlap and from the background a-particles. Secular equilibrium is also assumed, yet 234U in-grows during the time periods of the experiments, which of necessity have to be long enough to collect statistically large sets of counting data. And the 235U half-life is ultimately determined from the determined 238U half-life by assuming the 238U/235U ratio is constant, which is also crucial in every U-Pb age calculation. Yet significant variations in this crucial 238U/235U ratio have now been measured in all the rocks, accessory U-bearing minerals, and meteorites that are routinely U-Pb dated. Also, clearly observable trends of decreasing 238U and 235U half-life values were obtained from the direct counting experiments between 1932 and 1974. Such experiments should be given the most weight in determining the 238U and 235U half-lives, because in them the numbers of parent 238U and 235U atoms that decay over given time periods are directly counted. Yet to admit that the 238U and 235U decay rates may not have been constant in recent decades is tantamount to admitting that the 238U and 235U half-lives might never be determined precisely. Furthermore, since the 87Rb, 176Lu, 187Re, 147Sm, and 40K half-lives have all been determined by cross-calibration with the 238U half-life by forced agreement of Rb-Sr, Lu-Hf, Re-Os, Sm-Nd, K-Ar, and Ar-Ar ages respectively with U-Pb ages obtained for the same rocks, minerals and meteorites, none of these decay half-lives are really known accurately. Therefore, without accurately known decay half-lives, all radioisotope ages cannot be accurately determined or be considered absolute ages. Thus, all these radioisotope dating methods cannot be used to reject the young-earth creationist timescale, especially as current radioisotope dating methodologies are at best hypotheses based on extrapolating current measurements and observations back into an assumed deep time history for the cosmos. Instead, the actual observable experimentally-determined radioisotope decay data suggest that radioisotope decay rates have been decreasing in recent decades. This is consistent with the several lines of impeccable evidence that radioisotope decay rates were grossly accelerated during the year-long biblical global Flood cataclysm, and then the decay rates decelerated. That we may still be detecting the radioisotope decay rates decelerating is likewise consistent with the Flood occurring only about 4300 years ago.
Keywords: radioisotope dating, decay constants, half-lives, uranium-238, 238U, uranium-235, 235U, α-decay, lead-206, 206Pb, lead-207, 207Pb, uranium-lead dating, lead-lead dating, 238U/235U ratio, direct counting experiments, ion chambers, proportional counters, mass spectrometers, critical reviews, reevaluations, geological comparisons
Introduction
Radioisotope dating of rocks and meteorites is perhaps the most potent claimed proof for the supposed old age of the earth and the solar system. The absolute ages provided by the radioisotope dating methods provide an apparent aura of certainty to the claimed millions and billions of years for formation of the earth’s rocks. Many in both the scientific community and the general public around the world thus remain convinced of the earth’s claimed great antiquity.
However, accurate radioisotopic age determinations require that the decay constants of the respective parent radionuclides be accurately known and constant in time. Ideally, the uncertainty of the decay constants should be negligible compared to, or at least be commensurate with, the analytical uncertainties of the mass spectrometer measurements entering the radioisotope age calculations (Begemann et al. 2001). Clearly, based on the ongoing discussion in the conventional literature this is still not the case at present. The stunning improvements in the performance of mass spectrometers during the past four or so decades, starting with the landmark paper by Wasserburg et al. (1969), have not been accompanied by any comparable improvement in the accuracy of the decay constants (Begemann et al. 2001; Steiger and Jäger 1977), in spite of ongoing attempts (Miller 2012). The uncertainties associated with direct half-life determinations are, in most cases, still at the 1% level, which is still significantly better than any radioisotope method for determining the ages of rock formations. However, even uncertainties of only 1% in the half-lives lead to very significant discrepancies in the derived radioisotope ages. The recognition of an urgent need to improve the situation is not new (for example, Min et al. 2000; Renne, Karner, and Ludwig 1998). It continues to be mentioned, at one time or another, by every group active in geo- or cosmochronology (Schmitz 2012).
From a creationist perspective, the 1997–2005 RATE (Radioisotopes and the Age of The Earth) project successfully made progress in documenting some of the pitfalls in the radioisotope dating methods, and especially in demonstrating that radioisotope decay rates may not have always been constant at today’s measured rates (Vardiman, Snelling, and Chaffin 2000, 2005). Yet much research effort remains to be done to make further inroads into not only uncovering the flaws intrinsic to these long-age dating methods, but towards a thorough understanding of radioisotopes and their decay during the earth’s history within a biblical creationist framework.
One crucial area the RATE project did not touch on was the issue of how reliable have been the determinations of the radioisotope decay rates, which are so crucial for calibrating these dating “clocks.” Indeed, before this present series of papers (Snelling 2014a, b; 2015a, b; 2016) there have not been any attempts in the creationist literature to review how the half-lives of the parent radioisotopes used in long-age geological dating have been determined and to collate all the determinations of them reported in the literature to discuss the accuracy of their currently accepted values. After all, accurate radioisotope age determinations depend on accurate determinations of the decay constants or half-lives of the respective parent radioisotopes. The reliability of the other two assumptions these supposed absolute dating methods rely on, that is, the starting conditions and no contamination of closed systems, are unprovable. Yet these can supposedly be circumvented somewhat via the isochron technique, because it is independent of the starting conditions and is claimed to be sensitive to revealing any contamination, which is still significantly better than any radioisotope method for determining the ages of rock formations. Data points that do not fit on the isochron are simply ignored because their values are regarded as due to contamination. That this is common practice is illustrated with numerous examples cited from the literature by Faure and Mensing (2005) and Dickin (2005). On the other hand, it could be argued that this discarding of data points which do not fit the isochron is arbitrary and therefore is not good science, because it is merely assumed the “aberrant” values are due to contamination rather than that being proven to be so. Indeed, in order to discard such outliers in any data set, one must establish a reason for discarding those data points which cannot be reasonably questioned.
In order to rectify this deficiency, Snelling (2014a, b; 2015a, b; 2016) has documented the methodology behind and history of determining the decay constants and half-lives of the parent radioisotopes 87Rb, 176Lu, 187Re, 147Sm, and 40K which are used as the basis for the Rb-Sr, Lu-Hf, Re-Os, Sm-Nd, K-Ar, and Ar-Ar long-age dating methods respectively. He showed that there is still some uncertainty in what the values for these measures of the 87Rb, 176Lu, and 40K decay rates should be, in contrast to the apparent agreement on the 187Re and 147Sm decay rates. This uncertainty is especially prominent in determinations of the 176Lu decay rate by physical direct counting experiments. Furthermore, the determined values of the 87Rb decay rate differ when Rb-Sr ages are calibrated against the U-Pb ages of either the same terrestrial minerals and rocks or the same meteorites and lunar rocks. Ironically it is the slow decay rates of isotopes such as 87Rb, 176Lu, 187Re, and 147Sm used for deep time dating that makes precise measurements of their decay rates so difficult. Thus it could be argued that direct measurements of their decay rates should be the only acceptable experimental evidence, especially because measurements which are calibrated against other radioisotope systems are already biased by the currently accepted methodology employed by the secular community in their rock dating methods. Yet, the 87Rb, 176Lu, 187Re, 147Sm, and 40K decay half-lives have all ultimately been calibrated against the U-Pb radioisotope systems. This is the case even for the 147Sm decay half-life whose accepted value has not changed since it was calibrated against the U-Pb dating of two meteorites in the 1970s, in spite of the fact that more recent thorough physical direct counting experiments suggest a higher value. Thus confidence in U-Pb radioisotope dating as the “gold standard” is very questionable, as there are now known measured variations in the 238U/235U ratio that are critical to that method (Brennecka and Wadhwa 2012; Hiess et al. 2012), as well as uncertainties as to the 238U and 235U decay rate values (Mattinson 2010; Schoene et al. 2006; Schön, Winkler, and Kutschera 2004; Villa et al. 2016).
Therefore, the aim of this contribution is to further document the methodology behind and history of determining the present decay constants and half-lives of the parent radioisotopes used as the basis for the long-age dating methods. We need to explore just how accurate these determinations are, whether there really is consensus on standard values for the half-lives and decay constants, and just how independent and objective the standard values are from one another between the different methods. Of course, it is to be expected that every long-lived radioactive isotope is likely to show similar variation and uncertainty in half-life measurements because these are difficult measurements to make. However, even small variations and uncertainties in the half-life values result in large variations and uncertainties in the calculated ages for rocks and minerals, and the question remains as to whether the half-life values for each long-lived parent radioisotope are independently determined. We continue here with determinations of the uranium-238 (238U) and uranium-235 (235U) decay rates, which are the basis for the U-Pb and Pb-Pb dating methods.
Uranium, 238U and 235U Decay, and the U-Pb Dating Methods
The decay of the uranium isotopes 238U and 235U to the stable lead isotopes 206Pb and 207Pb respectively is the basis for the most frequently used methods of radioisotope dating. These not only derive from the transformation of 238U and 235U to 206Pb and 207Pb respectively, but also derive from the time-dependent “evolution” of common lead 204Pb from the decay of the intermediate daughters of 238U and 235U, and from the resulting isotopic composition of the accumulating daughter He (helium). Age determinations of rocks based on the decay of U and resulting accumulation of Pb and He were first attempted in the early years of the twentieth century by Rutherford (1906) and Boltwood (1907). Subsequently, Holmes (1913) used chemical U-Pb and U-He dates to propose the first geological timescale based on radioisotope dating in his book on the age of the earth.
The invention of the first mass spectrometer by Thomson (1911) was followed by the work of Dempster (1918) and Ashton (1919), who designed the mass spectrographs which they used in subsequent years to discover the naturally occurring isotopes of most of the elements in the periodic table and to measure their masses and abundances. The design of mass spectrographs was further improved in the 1930s, but it was the mass spectrometers based on a design by Nier (1940) that made possible the measurement and interpretation of variations in the isotopic composition of certain elements in natural materials such as minerals and rocks. Modern mass spectrometers follow his design and achieve a high level of accuracy and reliability of operation which enable isotope ratios to be measured for radioisotope dating, such as that based on the isotopic composition of Pb due to the decay of U to Pb, but also on the isotope ratios of common Pb. As a result of continuing refinement of the analytical procedures and of the sophistication of the instrumentation, the U-Pb and Pb-Pb methods of radioisotope dating are now regarded as the most precise and accurate geochronometers for determining the ages of terrestrial and extra-terrestrial minerals and rocks.
Uranium geochemistry
Uranium is element 92 (Z = 92) and a member of the actinide series in which the 5f orbitals are progressively filled with electrons. It occurs naturally in the tetravalent oxidation state U4+ with an ionic radius of 1.05 Å. But under oxidizing conditions it forms the uranyl ion (UO22+) in which U has a valence of 6+. The uranyl ion forms compounds that are soluble in water, so U is a mobile element under oxidizing conditions. In contrast to U, Pb (Z = 82) is in period 6 and is a group 14 post-transitional metal. It is insoluble in water, but is a chalcophile element because it reacts with sulfur. It forms Pb2+ and Pb4+ ions with ionic radii of 1.32 Å and 0.91 Å respectively, so Pb ions cannot substitute for U ions in minerals, because the Pb2+ has the wrong ionic charge and the Pb4+ ion has a smaller ionic radius that the U4+ ion.
In the course of partial melting of the rocks in the earth’s mantle, U is concentrated in the liquid (melt) phase and thus becomes incorporated into the more silica-rich products (Faure and Mensing 2005; Rudnick and Gao 2005). Thus the progressive geochemical differentiation of the earth’s upper mantle has enriched the rocks of the earth’s continental crust in U compared to those of the upper mantle. At an average of 1.3 ppm U is the fifty-first most abundant element in the earth’s crust, whereas Pb is regarded as quite a common element in the earth’s crust with an average of 11 ppm (Rudnick and Gao 2005). The concentrations of U and Pb increase from basaltic rocks (0.5 ppm U and 4 ppm Pb) to granites (5 ppm U and 23 ppm Pb) (Faure and Mensing 2005). The concentrations of U in the common rock-forming silicate minerals are uniformly low, on the order of a few ppm or less. Instead, U occurs primarily in certain accessory minerals in which it is either a major constituent or replaces other elements. These minerals include uraninite, zircon, baddeleyite, monazite, apatite, and sphene (titanite).
238U and 235U decay
Uranium has three naturally occurring isotopes, 238U, 235U, and 234U, all of which are radioactive. 238U and 235U are the principal U isotopes that each parent a chain of radioactive daughters ending in stable isotopes of Pb. The decay of 238U gives rise to what is called the uranium series, which includes 234U as one of the intermediate daughters and ends in stable 206Pb (fig. 1). The decay of 238U to 206Pb can be summarized by the equation
where Q = 47.4 MeV per atom or 0.71 calories per gram per year (Wetherill 1966). Each atom of 238U that decays produces one atom of 206Pb by emission of eight α-particles and six β-particles. The parameter Q represents the sum of the decay energies of the entire series in units of millions of electron volts and calories of heat produced per gram per year. Several intermediate daughters in this series (fig. 1) undergo branched decay involving the emission of either an α-particle or a β-particle. The chain therefore splits into separate branches, but 206Pb is the stable end product of all possible decay paths.
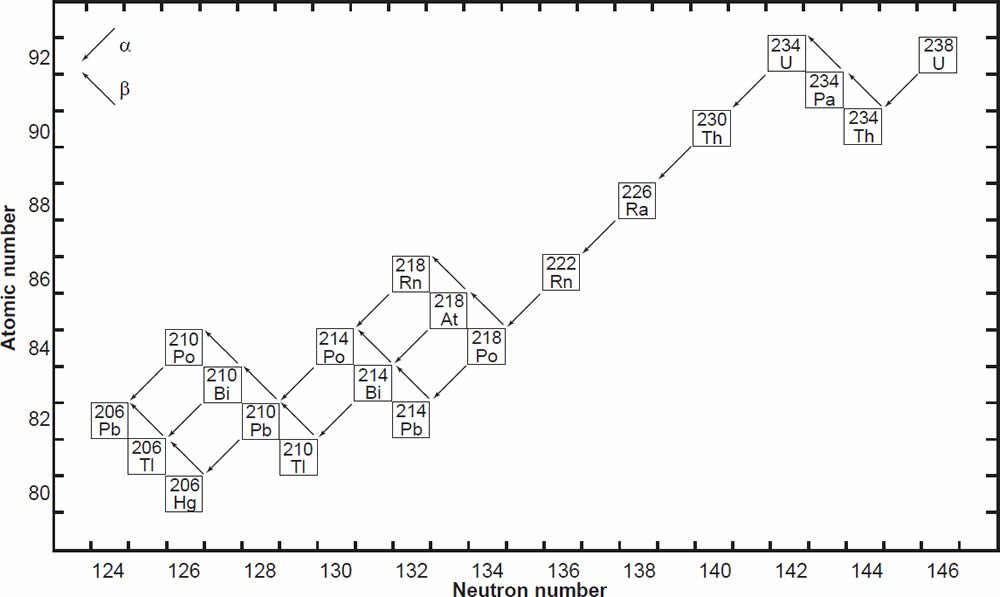
Figure 1. The decay chain of 238U resulting from the successive emission of α-particles and β-particles from intermediate isotopes as indicated (after Faure and Mensing 2005). The final decay product is stable 206Pb.
The decay of 235U gives rise to what is called the actinium series (fig. 2), which ends with stable 207Pb after emission of seven α-particles and four β-particles, as summarized by the equation
where Q = 45.2 MeV per atom or 4.3 calories per gram per year (Wetherill 1966). This series also branches as shown in Fig. 2.
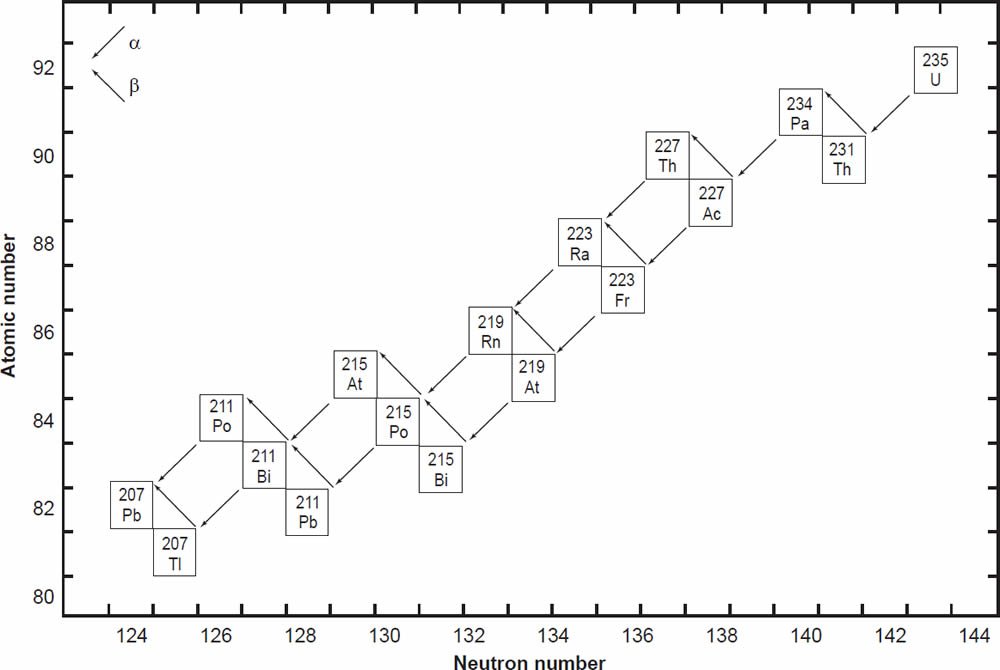
Figure 2. The decay chain of 235U resulting from the successive emission of α-particles and β-particles from intermediate isotopes as indicated (after Faure and Mensing 2005). The final decay product is stable 207Pb.
In spite of there being 33 isotopes of 12 elements formed as intermediate daughters in these two decay series (not counting 4He), none is a member of more than one series. In other words, each decay chain always leads through its unique set of intermediate isotopes to the formation of a specific stable Pb isotope. The decay of 238U always produces 206Pb, and 235U always produces 207Pb.
The half-lives of 238U and 235U are very much longer than those of their respective intermediate daughter isotopes. Therefore, these decay series satisfy the prerequisite condition for the establishment of secular equilibrium, provided none of the intermediate daughters escaped from the U-bearing mineral or were added from external sources (Faure and Mensing 2005) and sufficient time has elapsed. When secular equilibrium exists in a U-bearing mineral because it is a closed system, the decay rates of the intermediate daughters are equal to those of their respective parents, and thus the production rate of the stable daughter at the end of the decay chain is equal to the decay rate of its parent at the head of that chain. Therefore, the decay of 238U and 235U in minerals in which secular equilibrium has established itself can be treated as though it occurred directly to the respective 206Pb and 207Pb isotopes. As a result, the growth of these radiogenic Pb isotopes can be described by means of equations (1) and (2), which are similar to the equations used to represent the decay of 87Rb to 87Sr and 147Sm to 143Nd.
The U-Pb dating methods
The accumulation of stable daughter atoms from the decay of parent atoms over time is expressed by the equation known as the law of radioactivity, namely
where D* is the number of measured stable radiogenic daughter atoms, N is the number of measured parent atoms remaining, λ is the decay constant (decay rate), and t is the time since decay of the parent atoms began (Faure and Mensing 2005). Since D* and N can be measured in a mineral, then if λ is known the equation can be solved for t, which is thus declared to be the age of the mineral. Thus the accumulation of stable radiogenic 206Pb and 207Pb by decay of their respective parents 238U and 235U in a mineral is governed by equations derivable from equation (3) as follows
where λ1 and λ2 are the decay constants of 238U and 235U respectively; 238U/204Pb and 235U/204Pb are ratios of these isotopes calculated from the measured concentrations of U and Pb in the mineral; and the subscript i refers to the initial values of the 206Pb/204Pb and 207Pb/204Pb ratios.
To date U-bearing minerals by the U-Pb methods, the concentrations of U and Pb are measured by an appropriate analytical technique (usually isotope dilution), and the isotopic composition of Pb is determined by using a solid-source mass spectrometer, an ion-probe mass spectrometer, or an ICP mass spectrometer. The U-Pb dates are calculated by means of equations (4) and (5) being solved for t using assumed values of the initial isotope ratios of Pb (for example, Ludwig 1993) as follows
These are known as 206Pb and 207Pb model ages respectively. They are independent of each other, but will be concordant (that is, agree with each other) if the mineral samples satisfy the conditions for dating (Faure and Mensing 2005):
- The mineral has remained closed to U and Pb, and all the intermediate daughters throughout its history;
- Correct values are used for the initial Pb isotope ratios;
- The decay constants of 238U and 235U are known accurately;
- The isotopic composition of U is normal and has not been modified by isotope fractionation or by occurrence of a natural chain reaction based on induced fission of 235U; and
- All analytical results are accurate and free of systematic errors.
The assumption that the samples being dated remained closed to U, Pb, and all intermediate daughters throughout their history “is satisfied only in rare cases because U is a mobile element in oxidizing environments and therefore tends to be lost during chemical weathering” (Faure and Mensing 2005, 219, emphasis theirs). It is clearly hard to believe that this assumption is ever really satisfied over the claimed millions or billions of years. In addition, the emission of α-particles causes radiation damage to the crystal structures of the U-hosting minerals, which facilitates the loss of Pb and the other intermediate daughters in both decay chains. Consequently, U-Pb dates for rocks and minerals are rarely concordant, so procedures have been devised to overcome that problem.
The choice of the initial Pb isotope ratios would seem to only be a problem for dating rocks and minerals that have low U/Pb ratios and additionally are young. The numerical values of the initial Pb isotope ratios do not appear to significantly affect the calculated U-Pb ages of Precambrian rocks and minerals having high U/Pb ratios because their present Pb isotope ratios in most cases reach large values. Of course, this is only true if one assumes that little or no 206Pb or 207Pb was originally present in those rocks.
The decay constants and half-lives of 238U and 235U were fixed by the International Union of Geological Sciences (IUGS) Subcommission of Geochronology in 1975 (Steiger and Jäger 1977). At the same time a value of 137.88 was adopted for the 238U/235U ratio. Since then these values have been used in almost all U-Pb age calculations so as to avoid any potential confusion by the use of different values. It has been continually claimed that the numerical values of the 238U and 235U decay constants and half-lives are probably more accurately known that those of other long-lived radionuclides because of their importance in the nuclear industry. Therefore, refractory U-bearing minerals such as zircon (ZrSiO4) that often yield concordant U-Pb ages have been used to refine (that is, adjust) the decay constants of other radionuclides used in geochronology (Begemann et al. 2001; Snelling 2014a, b; 2015a, b; 2016).
It should be mentioned here that decay rates are not just measured and expressed by the parameter known as the decay constant (λ), but also by the parameter called the half-life (t½). The decay constant can be defined as the probability per unit time of a particular nucleus decaying, whereas the half-life is the time it takes for half of a given number of the parent radionuclide atoms to decay. The two quantities can be almost used interchangeably, because they are related by the equation:
Additionally, the issue of the abundances of the U isotopes and thus the adopted value of the 238U/235U ratio deserves further comment here. Real differences in the isotopic composition of terrestrial and extraterrestrial U have been reported. For example, uranium deposits of Precambrian age at Oklo in Gabon, Africa were found to be significantly depleted in 235U because the 235U appears to have been consumed by neutron-induced fission when these high-grade uranium deposits apparently became natural fission reactors at 1.8 Byr ago (Cowan 1976; Kuroda 1982; Lancelot, Vitrac, and Allègre 1975). The abundance of 235U in the ore mined at Oklo was as low as 0.3%, compared to 0.725% in normal U. Thus the 238U/235U ratio is as high in the Oklo U ore as 333.33 compared to the normal 137.88. However, natural fission reactors appear to be rare. Therefore, until recently there has been no compelling evidence to the contrary, so age determinations of terrestrial and lunar rocks and minerals, and of meteorites and their minerals, by the U-Pb method have been, and continue to be, based on a value of 137.88 for the present-day 238U/235U ratio. It is claimed that the effect of Pb loss on U-Pb dates can be minimized by calculating a date based on the 207Pb/206Pb ratio which is supposed to be insensitive to recent Pb loss provided that the Pb which was lost from the mineral had the same isotopic composition as the Pb which remained, that is, there has been no isotopic fractionation. The relationship between the 207Pb/206Pb ratio and time results from the difference in the half-lives of 238U and 235U. The desired equation is obtained by combining equations (4) and (5) above:
This equation has several interesting properties (Faure and Mensing 2005):
It involves the 235U/238U ratio which at 1/137.88 is regarded as a constant for all U of normal isotopic composition on and in the earth, the moon, Mars, and meteorites at the present time.
The equation does not require knowledge of the concentrations of U and Pb and involves only isotope ratios of Pb.
-
The left hand side of equation (9) is equal to the 207Pb/206Pb ratio of radiogenic Pb:
(10)where the asterisk * identifies the radiogenic isotopes.
Equation (9) cannot be solved for t by algebraic means because it is transcendental, but it can be solved by iteration and by interpretation in a table.
A difficulty arises in the solution of equation (9) when t = 0, because it yields the indeterminate result 0/0. This difficulty is overcome by means of l’Hôpital’s rule (Faure and Mensing 2005), which requires that the differentiated functions are differential over the entire open interval in question, that is, over millions to billions of years. Applying this rule, the value of (207Pb/206Pb)* at the present time (t = 0) is
Equation (11) indicates that the (207Pb/206Pb)* which forms by the decay of 238U and 235U at the present time is equal to the rates of decay of these two U isotopes at the present time. Substituting into equation (11) the relevant values for the 235U/238U ratio, and the decay constants λ1 and λ2 yields a value at the present time (t = 0) for (207Pb/206Pb)* of 0.04604. (Parenthetically, this procedure thus predicts a 207Pb/206Pb ratio of 0.04604 at the creation of the universe.)
The numerical values of (eλ1t − 1) and (eλ2t − 1) are listed in Table 1 and yield the (207Pb/206Pb)* ratios for increasing values of t ranging from t = 0 to t = 4.6 Byr. This table can be used to solve equation (9) for t by linear interpolation based on the (207Pb/206Pb)* ratio calculated from equation (10). Conversely, by determining the (207Pb/206Pb)* ratio in a mineral from measurements of its Pb isotope ratios, the age (t) of the mineral can be calculated by linear interpolation between the (207Pb/206Pb)* ratio values in Table 1. This is known as the 207Pb-206Pb model age.
| t, × 109 y | eλ1t − 1 | eλ2t − 1 | 207Pb*/206Pb |
|---|---|---|---|
| 0 | 0.0000 | 0.0000 | 0.04604 |
| 0.2 | 0.0315 | 0.2177 | 0.05012 |
| 0.4 | 0.0640 | 0.4828 | 0.05471 |
| 0.6 | 0.0975 | 0.8056 | 0.05992 |
| 0.8 | 0.1321 | 1.1987 | 0.06581 |
| 1.0 | 0.1678 | 1.6774 | 0.07250 |
| 1.2 | 0.2046 | 2.2603 | 0.08012 |
| 1.4 | 0.2426 | 2.9701 | 0.08879 |
| 1.6 | 0.2817 | 3.8344 | 0.09872 |
| 1.8 | 0.3221 | 4.8869 | 0.11004 |
| 2.0 | 0.3638 | 6.1685 | 0.12298 |
| 2.2 | 0.4067 | 7.7292 | 0.13783 |
| 2.4 | 0.4511 | 9.6296 | 0.15482 |
| 2.6 | 0.4968 | 11.9437 | 0.17436 |
| 2.8 | 0.5440 | 14.7617 | 0.19680 |
| 3.0 | 0.5926 | 18.1931 | 0.22266 |
| 3.2 | 0.6428 | 22.3716 | 0.25241 |
| 3.4 | 0.6946 | 27.4597 | 0.28672 |
| 3.6 | 0.7480 | 33.6556 | 0.32634 |
| 3.8 | 0.8030 | 41.2004 | 0.37212 |
| 4.0 | 0.8599 | 50.3878 | 0.42498 |
| 4.2 | 0.9185 | 61.5752 | 0.48623 |
| 4.4 | 0.9789 | 75.1984 | 0.55714 |
| 4.6 | 1.0413 | 91.7873 | 0.63930 |
Although U occurs in a large number of minerals, only a few are suitable for dating by the U-Pb methods. To be useful for dating, a mineral must be retentive with respect to U, Pb and the intermediate daughters, and it should be widely distributed in a variety of rocks. The minerals that satisfy these conditions include zircon, baddeleyite, monazite, apatite, and sphene (titanite). All of these minerals contain trace amounts of U but low concentrations of Pb, giving them high U/Pb ratios favorable for dating. For example, concentrations of U in zircons range from a few hundred to a few thousand parts per million and average 1350 ppm (Faure and Mensing 2005). The presence of U in zircon is due to the isomorphous substitution within the zircon crystal lattice of U4+ (ionic radius 1.05 Å) for Zr4+ (0.87 Å), though this substitution is limited by the differences in their ionic radii. However, whereas U4+ is admitted into zircon crystals, Pb2+ is claimed to be excluded because of its large ionic radius (1.32 Å) and its low charge (2+). Therefore, zircons are supposed to contain very little Pb at their time of formation and have high U/Pb ratios. This appears to enhance their sensitivity as a geochronometer, so zircons have for several decades become increasingly used for dating via the U-Pb methods.
The several assumptions involved in the various U-Pb and Pb-Pb model and isochron dating methods have a somewhat tenuous validity, because they are based on an unknown and unconfirmed uniformitarian evolutionary past history. Thus Snelling (2000) has extensively documented with many examples from the literature of the numerous failures of these radioisotope dating methods because of inheritance and contamination, contrary to two of three underlying assumptions involved in the methods. Yet in spite of that, all these methods depend on the decay constants or half-lives of 238U and 235U being known accurately. Furthermore, the isotopic composition of U has to have been normal and must not have been modified by isotope fractionation or by occurrence of a natural chain reaction based on induced fission of 235U, that is, the 238U/235U ratio has to be known and must have remained constant at that value. So it is to the determinations of these crucial values we now turn.
238U and 235U Decay Half-Life Determination Methods
Three approaches have been used since 1932 to determine the decay constants and half-lives of the long-lived radioactive 238U and 235U isotopes—direct counting, geological comparisons, and critical review and re-evaluation of previous experimental determinations by both direct counting and geological comparisons.
Direct counting
In this technique, experiments simply require the determination of the α-emission rate from a known weight of isotope. The α-activity is counted using various appropriate instruments, and then divided by the total number of radioactive atoms in the sample used. In practice, determining the weights of samples requires:
- chemical analysis of the amount of uranium present,
- tests for the presence of other interfering elements,
- mass spectrometric analysis to determine the fraction of uranium present in the form of the desired isotope, and
- a sample-preparation technique which ensured that each sample contained an accurately known weight.
Determining the α-emission rate requires that
- each sample be counted with a precisely known counting efficiency,
- each sample be counted long enough to make negligible the overall statistical counting error, and
- an energy analysis be made to determine the fraction of the total α-activity derived from the isotope of interest (although an alpha spectrometer should not need an energy analysis with properly prepared samples).
Among the difficulties of this approach are the self-shielding of finite-thickness solid samples, the low specific α-activities, imprecise knowledge of the isotopic composition of the parent element, the detection of very low-energy α-decays, and problems with detector efficiencies and geometry factors (Begemann et al. 2001).
238U decay rate
A review of the literature reveals that only nine direct counting experiments to determine the 238U decay rate have been performed since the first in 1932 (Kovarik and Adams 1932). Furthermore, the last of those nine experiments was in 1971 (Jaffey et al. 1971), and none have been reported in the 45 years since then. Thus the Jaffey et al. (1971) value of the 238U half-life remains the standard still currently used in geochronology (Schmitz 2012; Villa et al. 2016). In the last 45 years any refinements to this directly counted value of the 238U half-life have been by critical re-evaluation of the corrected data from all nine early (1932–1971) experiments, or by geological comparisons.
In the nine direct counting experiments to determine the 238U decay rate various samples were used. Most used what was described as “natural uranium” (Curtiss, Stockman, and Brown 1941; Kienberger 1949; Kovarik and Adams 1932, 1955; Schiedt 1935; Steyn and Strelow 1960), presumably in the form of U3O8. However, in the case of the Curtiss, Stockman, and Brown (1941) experiment it began with a uranium compound being sprayed onto a platinum disk which was ignited to glowing in a bunsen burner flame to presumably produce the U3O8. Other experimenters also used other uranium compounds such as uranium nitrate (Jaffey et al. 1971; Leachman and Schmitt 1957) or “depleted uranium” (Kienberger 1949; Leachman and Schmitt 1957).
Furthermore, the different experimenters used different sample preparation procedures. Kovarik and Adams (1932, 1955) prepared their two samples by slow settling of very fine particles of U3O8 out of chloroform onto aluminum plates. The plates were weighed before coating and subsequently when the thin U3O8 films had dried. The average thicknesses or surface densities of the U3O8 films were 0.4 and 1.6 mg U3O8 per cm2. In contrast, Schiedt (1935) electroplated two samples of “natural uranium” on plates with a surface density of 1 mg per cm2 for measurement of their specific activities, before being dissolved and chemically analyzed. However, as noted already, Curtiss, Stockman, and Brown (1941) sprayed a “uranium compound” onto a platinum disk and then ignited it until glowing in a bunsen burner, presumably to U3O8, and weighed to determine the average surface density before measuring the α-activity. And yet in a different procedure, Kienberger (1949) prepared samples of both natural uranium and “highly depleted uranium” by electroplating known weights of uranium before measuring the specific activities and subsequently analyzing for residual uranium in the residual solutions as a cross-check. On the other hand, the measurement of the 238U half-life was incidental in the experiment by Leachman and Schmitt (1957), who used depleted 238U as uranium nitrate samples painted onto platinum plates, which were ignited to U3O8 and weighed to determine an average surface density of 0.2 mg per cm2 before the α-activities were measured. Steyn and Strelow (1960) dissolved samples of natural uranium in an organic liquid scintillator and the β-active daughters separated from the solutions (which would have been a difficult measurement even with today’s technology). Finally, Jaffey et al. (1971) used weighed aliquots of aqueous stock solutions of uranyl nitrate (presumably, but not stated, traceable to the National Institute of Standards) whose chemical purity was checked, that were sealed in polyethylene ampoules. The ampoules were weighed before and after filling to determine the exact weight of each aliquot. Weighed aliquots had isopropyl alcohol added to them before being transferred to a cell in which the uranium was molecular-plated onto aluminum plates so that the surface density of 238U per sample varied from 0.3 to 0.5 mg per cm2. Two different batches of depleted 238U whose isotopic compositions were first determined by mass spectrometry analyses, then had their α-activities counted.
Different instruments were used in these experiments for the direct counting of α-activities to determine the 238U decay rate (half-life). Curtiss, Stockman and Brown (1941), Kienberger (1949), Kovarik and Adams (1932, 1955), Leachman and Schmitt (1957), and Schiedt (1935) all used ion chambers, although they used various arrangements and alignments of the equipment. Ion chambers cannot distinguish between beta, gamma, or alpha radiation and therefore cannot produce an energy spectrum of the incident radiation. Kovarik and Adams (1932, 1955) used a carefully drilled plate of collimator holes over their samples, so that each hole, in effect, acted as a low-geometry counter whose geometry factor could be calculated. Thus back-scattering and sample self-absorption were apparently eliminated. However, Schiedt (1935) used an ion chamber (α-particle counter) with “intermediate geometry,” whereas Curtiss, Stockman, and Brown (1941), Kienberger (1949), and Leachman and Schmitt (1957) all used instruments with 2π geometry, the solid counting angle. In each experiment the α-particle back-scattering and self-absorption corrections needed to be calculated and factored into the determinations and their uncertainties. Such corrections, while feasible, are difficult to make to the desired accuracy.
In contrast, Steyn and Strelow (1960) used a liquid scintillator with 4π geometry (a wider solid counting angle), while Jaffey et al. (1971) used a proportional counter with intermediate geometry (fig. 3). Jaffey et al. (1971) went to great lengths to describe in detail the instrument they used and why they used it. They argued that the back-scattering and self-absorption corrections are unnecessary when the measured α-particles are restricted to those emitted at fairly large angles to the surface of the sample plate. This is due to the fact absorption in the uniformly deposited, molecular-plated samples they used occurs only for α-particles emitted at small angles to the samples’ surfaces, while back-scattered α-particles, arising as they do from multiple electronic scattering, are also a relatively small angle phenomenon (Jaffey at al. 1971). Furthermore, the counter aperture (KK in fig. 3) needed to be large relative to the diameter of the sample deposit (on the sample mount H in fig. 3), in order to keep the detector geometry reasonably constant over the sample surface. Thus a counter of fairly large dimensions (M in fig. 3) was also required so as to make the geometry less sensitive to slight errors in measuring the thickness of the sample mount. In essence, this was all done so that the sample-detector configuration would approximate a point source and thus eliminate the need for complex geometric corrections to the counting geometry.
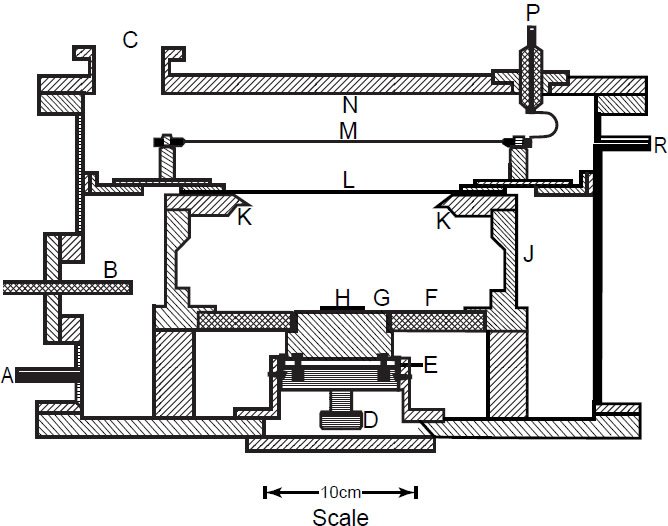
Figure 3. The diagrammatic configuration of the intermediate-geometry proportional α-counter used by Jaffey et al. (1971). A and R = the gas inlet and outlet respectively for the flowing argon (10% methane); C = vacuum pump; G = sample support (precisely positioned); H = sample mount (centered on sample support); K = accurately machined circular aperture (with a 0.001-inch-thick edge and precisely measured diameter); L = thin plastic film with an evaporated-gold conducting layer (~ 0.6 mg/cm2); M = proportional-counter wires (spanning the circular area); and P = high voltage and signal lead.
In each of these experiments the specific activity of 238U in the α-energy spectrum of the uranium sample was measured to determine the 238U half-life. As can be seen in Fig. 4, while the 238U peak dominates the α-energy spectrum, the pulse height is not easy to measure given the breadth of the peak, the extent of the peak tails, and the contributions from the other U isotopes. Jaffey at al. (1971) provided an analysis of how they dealt with pulse pile-up where counts were high and during the dead times of the counters. They estimated the uncertainty due to dead-time losses was <0.005%. In any case, it is normally assumed that the α-activity contribution of 238U to the total α-activity of freshly separated uranium samples is 48.875% (assuming secular equilibrium). To eliminate interferences and thus separate and increase the 238U pulse height, samples highly enriched in 238U were sometimes used, but as can be seen in Fig. 5 there can still be a long significant tail to the 238U peak which can affect the determination of the α-activity specific to 238U. At no time did these experimenters cross-calibrate their instrument using National Institute of Standards uranium samples, although they did use National Bureau of Standards (NBS) uranium oxide samples to cross-check their preparation procedure of their samples for α-counting.
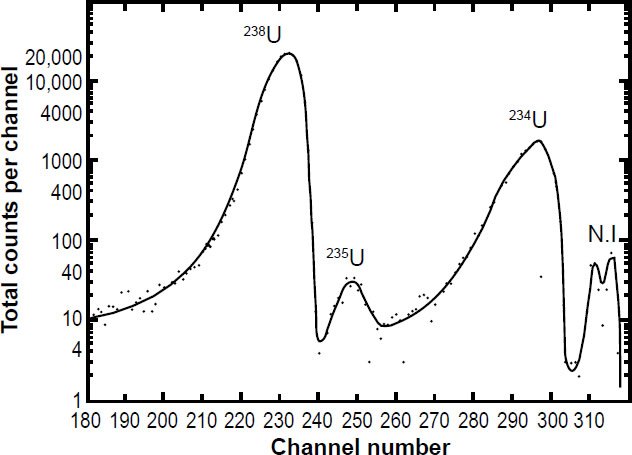
Figure 4. Energy pulse height analysis of α-particles from a highly enriched 238U sample obtained using an intermediate geometry proportional counter (fig. 3) and a large-area silicon-junction surface-barrier detector obtained by Jaffey et al. (1971) from which they extracted the specific 238U α-activity to determine the 238U half-life. The peak labeled N.I. was not identified, but contributed only about 0.1% of the α-activity.
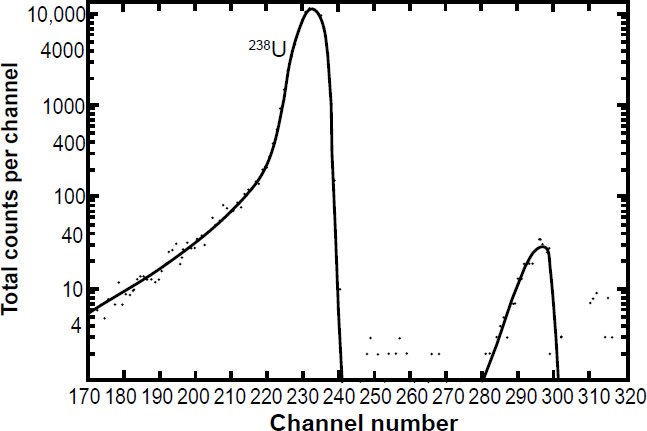
Figure 5. Energy pulse height analysis of α-particles from a second highly enriched 238U sample obtained using an intermediate geometry proportional counter (fig. 3) and a large-area silicon-junction surface-barrier detector obtained by Jaffey et al. (1971) from which they extracted the specific 238U α-activity to determine the 238U half-life.
The fact that no further experimental determinations of the 238U half-life by direct counting have been made in the 45 years since Jaffey et al. (1971), and the fact that the 238U half-life value they determined is still the recommended value for use in geochronology (Villa et al. 2016), is an amazing testimony to the skill and thoroughness of these experimenters and the caliber of the counting instrument and experimental procedures they used. All the details of their equipment and their counting procedures are carefully elaborated in their paper. The errors quoted by Jaffey et al. (1971) were statistical (standard error of the mean), based on the observed scatter of the data, which exceeded that expected from counting statistics alone. They believed, as stated in their abstract, that if present any systematic errors would no more than double the errors they quoted. They never explicitly gave their reasons for this statement, although they can be deduced from their lengthy discussion assessing errors and uncertainties at each step in their experimental procedure. The accuracy of their error assessment was subsequently extolled by Schön, Winkler, and Kutschera (2004), who stated that the several sources of systematic uncertainties were treated in detail by Jaffey et el. (1971), and that because the uncertainty in the 238U half-life value determined by Jaffey et el. (1971) was essentially based on their statistical consistency tests employing various samples, their conclusions were found to be well warranted. Thus Schön, Winkler, and Kutschera (2004) doubled the uncertainty in their evaluation of the 238U half-life value, based on that statement by Jaffey et al. (1971), and this has been subsequently agreed with by Villa et al. (2016). Yet it is incredible that a precision alpha spectrometer with monolayer deposited samples has not been used subsequent to the Jaffey et al. (1971) experiments to measure this critical decay rate constant, since the necessary technology has existed since at least the late 1970s.
235U decay rate
A review of the literature indicates that 11 direct counting experiments to determine the 235U decay rate have been performed since the first in 1949 (Kienberger 1949). The last such experiment was in 1993 (Bueno and Santos 1993). In the 23 years since then any refinements to the directly counted value of the 235U half-life have been by critical re-evaluation of the corrected data from all 11 (1949–1993) experiments, or by geological comparisons. Yet the 235U half-life value determined by Jaffey et al. (1971) still remains the value used by convention in geochronology (Villa et al. 2016).
In the 11 direct counting experiments to determine the 235U decay rate various samples were used. Six used what was described as “natural uranium” (Clark, Spencer-Palmer and Woodward 1957; Deruytter, Schroder, and Moore 1965; Kienberger 1949; Sayag 1951; Würger, Meyer and Huber 1957), presumably in the form of U3O8. On the other hand, four experiments used what was described as “enriched uranium” (Beuno and Santos 1993; Fleming, Ghiorso, and Cunningham 1952; Knight 1950; White, Wall, and Pontet 1965) and two experiments used “highly enriched uranium” (Deruytter and Wegener-Penning 1974; Jaffey et al. 1971). In most experiments it is not clear how the uranium samples used were enriched in 235U. However, Beuno and Santos (1993) used material from recycled fuel elements containing ≈ 20% 235U.
The sample preparation procedures used in each experiment are also not clear, but it appears that in many of the experiments carefully weighed sample solutions were electroplated onto sample mounting plates that were then placed in the instruments used. The sample preparation procedure used by Jaffey et al. (1971) has already been described above. Of course, in all these experiments the mass concentrations of the 238U and 235U isotopes in the sample solutions and thus in the electroplated samples had to be determined by independent procedures. In the experiments performed by Jaffey et al. (1971) that procedure involved titrations with solutions of acids and further weighing of the end products, whereas White, Wall, and Pontet (1965) weighed the sample plates then used spectrophotometry or coulometry and back-to-back fission counting after α-counting of the samples. Other experimenters used mass spectrometers for these isotopic mass determinations.
Different instruments were also used in these experiments for the direct counting of α-activities in the samples to determine the 235U decay rate (half-life). Clark, Spencer-Palmer, and Woodward (1957), Fleming, Ghiorso, and Cunningham (1952), Kienberger (1949), Knight (1950), Sayag (1951), and Würger, Meyer, and Huber (1957) all used ion chambers, although they used various arrangements and alignments of the equipment. It should be noted that ion chambers are primarily used to count β and γ radiation, but they can be used to count α-particles with the proper precautions. Fleming, Ghiorso, and Cunningham (1952) used an ion chamber (as an α-particle counter) with “intermediate geometry” of measured dimensions, whereas Kienberger (1949) used an ion chamber with 2π geometry. On the other hand, Deruytter, Schroder, and Moore (1965), Deruytter and Wegener-Penning (1974), and White, Wall, and Pontet (1965) all used solid-state detectors, which in most experiments were Si-solid-state-junction detectors. As already described above, Jaffey et al. (1971) used a proportional counter with intermediate geometry (fig. 3), which they went to great lengths to describe in detail and why they used it. And then in an entirely different approach, Beuno and Santos (1993) used a gas scintillator for the α channel and a NaI (Tl-doped) scintillator for the γ channel.
The 235U half-life measurements were not easy to make in these experiments because in the energy spectrum analyses the 235U peak is secondary to the 238U peak (fig. 6). It is necessary to measure the 235U peak in order to ascertain the α-activity specific to 235U per unit time to obtain the 235U decay rate independently from the dominant 238U α-activity and decay rate. That is why Bueno and Santos (1993) used two different scintillators, so they could measure the specific activity of 235U by the α-γ coincidence from the respective channels obtained from respective scintillators. The gas scintillation proportional counter was used to measure the 235U specific α-activity of their 235U-enriched U3O8 sample source, while the NaI (Tl-doped) solid-state scintillator simultaneously measured coincidently the γ-radiation specific to 231Th, the immediate decay product of 235U in the same 235U-enriched U3O8 sample source (see fig. 2). On the other hand, Fleming, Ghiorso, and Cunningham (1952), Knight (1950), White, Wall, and Pontet (1965), and Jaffey et al. (1971) all measured the specific α-activity of the 235U peak in the α-energy spectrum, whereas Kienberger (1949) determined the specific α-activity of 235U by subtracting the specific α-activity of the 238U peak. As can be seen in Fig. 7, Jaffey et al. (1971) enhanced the resolution of the specific α-activity of 235U by using a sample highly enriched in 235U (99.999%), which also increased the 235U pulse height and intensity (total counts per channel) relative to the background and any other peaks.
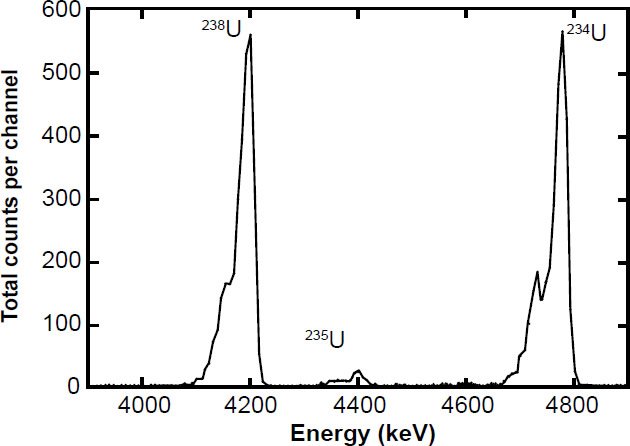
Figure 6. Alpha (α) energy spectrum from a natural uranium sample measured with an ion-implanted solidstate detector (after Schön, Winkler, and Kutschera 2004). Compared to the 238U and 234U peaks the pulse-height of the 235U peak is very small, which makes determination of its specific α-activity or the 235U/238U activity ratio difficult.
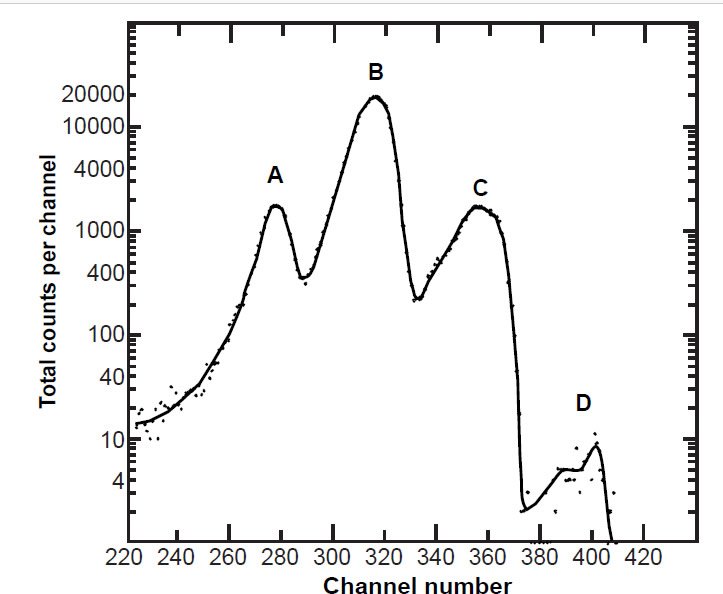
Figure 7. Energy pulse height analysis of α-particles from a highly enriched 235U sample obtained using an intermediate geometry proportional counter (fig. 3) and a large-area silicon-junction surface-barrier detector obtained by Jaffey et al. (1971) from which they extracted the specific 235U α-activity to determine the 235U half-life. The measured relative intensities of the major peaks were approximately A:B:C = 0.089:1.00:0.087, so they ascribed the B peak to the mixture of lines at 4.36–4.396 MeV providing the major 235U intensity (≈85% of the total 235U α-spectrum). The α-particles at D were ascribed to 234 U (or 233U).
Otherwise, Sayag (1951), Clark, Spencer-Palmer, and Woodward (1957), Würger, Meyer, and Huber (1957), and Deruytter, Schroder, and Moore (1965) all derived the specific α-activity of 235U from the activity ratio 235U/238U using the full α-energy spectrum. The 235U peak is dwarfed by the 238U and 234U peaks (fig. 6), and the total 235U α-spectrum actually consists of three peaks (fig. 7), the most significant (the so-called central peak which accounts for ≈85% of total 235U α-spectrum) lying in between and separated from the 238U and 234U peaks (see fig. 6 again). So determination of the specific 235U α-activity and thus the 235U half-life from the 235U/238U activity ratio depends on the estimate of the intensity (and associated uncertainty) of the central 235U peak, which in turn depends on the spectrum resolution, which is determined by the detector, the thickness and homogeneity of the sample, and the collimation of the detected α-particles (Schön, Winkler, and Kutschera 2004). Furthermore, because the experimenters used the 235U peak heights to determine their relative contributions to the overall 235U peak strength, and each peak height is differentially affected by energy straggling, this is a risky methodology especially for α-particles. In a variation of this methodology, Deruytter and Wegener-Penning (1974) also used a 99.999% 235U enriched sample to better determine the branching ratio of the dominant central 235U peak, that is, how much of the specific 235U α-activity is in the central peak compared to the two smaller side peaks (fig. 7), which in the full α-energy spectrum obtained from natural uranium disappear into the very large 238U and 234U peaks (fig. 6). To calculate accurately the intensity of the central 235U peak situated around 4.35 MeV required estimating the number of α-counts in the tailing on the low energy side of the central peak (that is, the fraction under the 4.20 MeV peak) and the number of α-counts in the central peak in the overlap region with the 4.5 MeV peak (fig. 8). However, peak extractions like this are seldom more accurate than ± 3%.
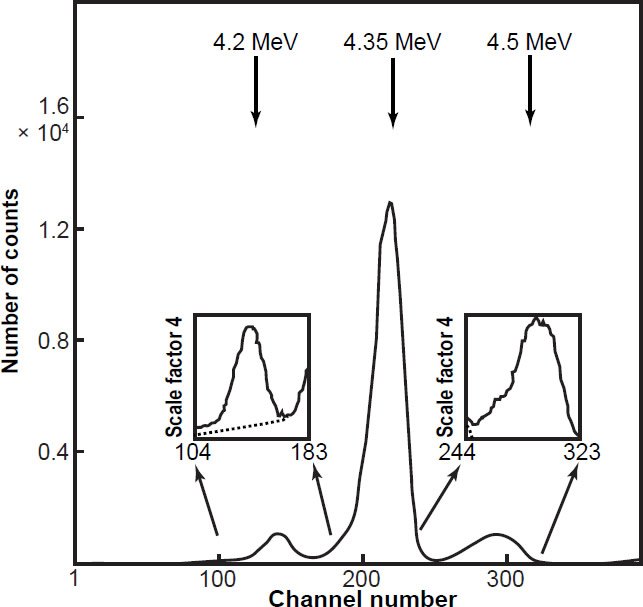
Figure 8. Experimental α-energy spectrum of a highly enriched 235U sample obtained by Deruytter and Wegener-Penning (1974) using a Si-solid-state-junction surface-barrier detector with low counting geometry. The dotted lines in the insets indicate the contributions of the central line (4.35 MeV) to the side peaks.
Judged from the fact that many of the earlier (pre-1960) direct counting experiments to determine the 238U and 235U half-lives yielded results that are not compatible with one another within the stated uncertainties, it would appear that not all the measurement uncertainties may have been accounted for, and therefore the stated uncertainties may be unrealistically small. According to Begemann et al. (2001) many of those experiments may thus have been plagued by unrecognized systematic errors. As the nature of these errors is obscure, it is not straightforward to decide which of the, often mutually exclusive, results of such direct counting experiments is closest to the true value. Yet it is intriguing that in spite of the determinations of the half-lives via geological comparisons and critical review reevaluations of all the experimental data since 1971 the geochronological community has still recommended the Jaffey et al. (1971) values (Schmitz 2012; Schön, Winkler, and Kutschera 2004; Villa et al. 2016). Furthermore, the presence of unknown systematic biases makes dangerous any averaging of all these direct counting experimental determinations. It is possible that reliable results of careful workers, listing realistic uncertainties, will not be given the weights they deserve—this aside from the question of whether it makes sense to average numbers that do not always agree within the stated uncertainties. In any case, there is a natural tendency for bias towards the most recent measurements as though the more “modern” equipment and methodologies guarantee better results, when in fact the earlier experimenters may have been more intimately involved and careful with their equipment and methodologies to obtain excellent results. Since the Jaffey et al. (1971) determined 238U and 235U half-lives are still the recommended values in use today, that at least discounts any overreliance by the early experimenters on computers in their direct counting methodologies. In contrast, the more recent efforts to better determine these half-life values via geological comparisons and critical review reevaluations of all the experimental data have heavily relied on computers, which has resulted in convergence of the recommended half-life values.
More accurate determinations still needed
As stated earlier, it is incredible that a precision alpha spectrometer with monolayer deposited samples has not been used subsequent to the Jaffey et al. (1971) experiments to measure these critical decay rate constants, since the necessary technology has existed since at least the late 1970s. Furthermore, Schön, Winkler, and Kutschera (2004) raised the question of whether these two uranium isotopes “will continue to provide the most accurate clocks for geochronology.” Indeed, they did not give recommended values for the 238U and 235U half-lives because they felt the weighted mean values and their uncertainties their critical review and reevaluation provided are obviously dominated by the results of Jaffey et al. (1971) and therefore essentially reproduced them. Yet even though the generally accepted Jaffey et al. (1971) half-life values are in agreement with other previous results, Schön, Winkler, and Kutschera (2004) regarded the situation as “far from satisfactory,” as in the intervening decades those values have simply not been confirmed with comparable accuracy. In their judgment, recommended values of the 238U and 235U half-lives would require at least two concordant results of comparable accuracy. Furthermore, even though they regarded the unweighted mean half-life values as more reliable within their uncertainties, they still miss the accuracy demanded by the geochronologists who use them, so they are thus of limited use. Indeed, based on the enormous increase in accuracy of mass-spectrometric measurements, geochronologists now consider the uncertainties of the 238U and 235U half-lives to be the limiting factor in their rock and mineral age determinations (Begemann et al. 2001; Renne, Karner, and Ludwig 1998).
Thus, because they realized the likelihood of mass-spectrometric techniques further improving (which they definitely have in the past decade), Schön, Winkler, and Kutschera (2004) urged that concerted efforts be made to re-measure the 238U and 235U half-lives with improved accuracy. They argued that chemical and mass-spectrometric expertise should be harnessed to specify and prepare highly enriched samples and be combined with the necessary expertise for activity measurements in order to achieve accuracies in the order of 0.1%. Yet realistically, any such determinations with an accuracy better than 1% is a tall order even with modern equipment. However, Schön, Winkler, and Kutschera (2004) suggested that α-XL and α-γ coincidence counting could be advantageously applied for decay-rate measurements on 238U and 235U samples, respectively. They suggested that high-efficiency detector configurations would have to be used in view of the low specific activities of about 12.5 Bq per mg (238U) and about 80 Bq per mg (235U). Furthermore, they thought that liquid-scintillation techniques should also be considered as appropriate counting methods, as already demonstrated in their view by Steyn and Strelow (1960) in determining the 238U half-life, and by Kossert and Günther (2004) in determining the 40K half-life.
Philosophical considerations
Begemann et al. (2001) had already made a “Call for an improved set of decay constants for geochronological use,” per the title of their paper. They had concluded that among all radioisotope dating systems in use, the U-Pb systems, and by inference the Pb-Pb system, are the least plagued by uncertainties. However, they realized that this is in large part because the counting experiment of Jaffey et al. (1971) has never been repeated with a claim for a comparable accuracy. Therefore, they conceded that there are no replicate experiments to average (weighted or unweighted), and thus the Jaffey et al. (1971) decay constants and half-lives are “gospel.”
Moreover, Begemann et al. (2001) admitted that ages based on the decay of uranium are used currently as standards to derive the decay constants and half-lives relevant to other radioisotope dating systems (as also concluded by Snelling 2014a, b; 2015a, b; 2016), but never vice versa. Because of this fact, there are by definition also no discrepancies between decay constants and thus half-lives derived from counting experiments and from such age comparisons.
To then say as they do that it is thus almost a philosophical question whether or not this calibration (nay forced) agreement between the rock-dating radioisotope half-lives should be changed by repeating the direct counting experiments is an admission that geochronologists have chosen not to do further direct counting determinations of these half-lives lest the results overturn their neat scheme of cross-calibrations against the accepted 238U half-life value. In other words, they admit that, because they tacitly assume the different parent radioisotopes should yield the same ages on the same rocks, minerals, or meteorites because all the radioisotope decay “clocks” in them must have all started at the same time zero when the rock, mineral or meteorite formed and then decayed (“ticked”) at the same rate through the assumed millions of years at today’s measured rate, it is legitimate to adjust the other half-lives so that their radioisotope ages agree with the U-Pb ages for the same rocks, minerals, and meteorites. Of course, Begemann et al. (2001) go on to acknowledge that there are some lingering doubts as to what the statement by Jaffey et al. (1971) that “systematic errors, if present, will no more than double the quoted errors” might have been based upon. Indeed, it is hard not to have serious doubts about the stated errors in most of these measurements; both random and systematic. Furthermore, an argument in favor of improving on the accuracy of the decay constants and half-lives of both 235U and 238U would still be that their uncorrelated uncertainties determine the finite width of the concordia curve (Ludwig 2000; Min et al. 2000), and that in turn has implications as to whether or not U-Pb ages are rigorously concordant.
Of course, Begemann et al. (2001) concede that all the relevant decay constants and half-lives are in need of improvements, especially those of 238U and 235U. They agree that ideally redeterminations should be attempted by repeating direct counting experiments, but then that should not be the only technique. They still favored cross-calibration with geological comparisons, even though they had already admitted that such age comparisons by their very philosophical nature eliminate the possibility of discrepancies. However, geological comparisons should not be considered in determining basic physical “constants” such as decay rates. They can really only be used as checks for these radioisotope dating methods. Of course, such attempted cross-calibrations by geological comparisons certainly would strengthen their “belief” in any new values and presumably make those new values more readily accepted by the geochronologists (Begemann et al. 2001). In needed further direct counting experiments they suggested it would be necessary to involve experimental research groups familiar with all intricacies of “absolute” counting. But then is it fair to ask whether such groups exist? Furthermore, the desired level of accuracy will hardly be attainable by single workers, or teams, occasionally dabbling in such counting experiments, and then with the results being a side effect of measurements with entirely different primary aims. It was the firm belief of Begemann et al. (2001) that only devoted experiments dedicated to the special purpose of improving the accuracy of the decay constants would bring the anticipated results. In such experiments they saw room for improvements by employing isotope dilution methods to determine the number of radioactive atoms involved in the experiments. Yet somehow unbiased NIST standards must come into play in calibrating the equipment used in such experiments, instead of the biased standards employed in the Ar-Ar dating method, and sometimes the U-Pb method.
Ironically, the more recent scathing “meta-analysis of geochronologically relevant half-lives” by Boehnke and Harrison (2014) appears to have been ignored by the geochronology community. Their “meta-analysis” used six criteria to assess the validity of any half-life determination study:
- It must be peer reviewed and widely accessible;
- It must describe the radioactive sample in detail, including weight, and elemental and isotopic compositions;
- It must describe the experimental apparatus in sufficient detail to assess potential sources of analytical error;
- It must yield results with an appropriate signal/ noise ratio;
- It must not be superseded by, or included in, later results from the same laboratory; and
- It must include measures of uncertainty derived from presented data.
What they also stated was that “decay constants are, literally, physical constants,” which was an admission of their bias that radioisotope decay rates cannot have varied in the past but must have always been constant.
Nevertheless, using these criteria Boehnke and Harrison (2014) found only four studies of the 235U decay rate (Deruytter and Wegener-Penning 1974; Fleming, Ghiorso, and Cunningham 1952; Jaffey et al. 1971; White, Wall, and Pontet 1965) and one study of the 238U decay rate (Jaffey et al. 1971) worthy of examination, that is, acceptable. Indeed, they were scathing of Schön, Winkler, and Kutschera (2004) in their review of experimental determinations for not applying objective criteria, and suggested that the paucity of reliable U decay studies is far more significant than previously expressed. Thus it is hardly surprising they echoed the call of Begemann et al. (2001) for new determinations of the 238U and 235U half-lives, and stated that a priority for the geochronological community must be support of new, high precision and accuracy measurements. Furthermore, they opined that despite the decay of U to Pb having grown to be the most widely used decay system for geochronology, the 238U and 235U decay half-lives have not been remeasured since Jaffey et al. (1971), although analytical capabilities have been vastly improved over the intervening 45 years. They concluded that the availability of highly enriched 235U and its short half-life relative to 238U suggests the potential for decay counting experiments of superior precision and accuracy. And until the 238U decay constant is remeasured through laboratory equipment, other geochronological systems (87Rb, 40K, 147Sm, etc.) should instead be calibrated to 235U rather than 238U! Yet they admitted that proposal would increase the 235U half-life uncertainty by more than 50% of that reported by Jaffey et al. (1971). Clearly, they stated, current knowledge of half-lives for many radiometric systems is the limiting parameter in achieving absolute age accuracies of better than ca. 1%. Furthermore, to truly achieve ± 0.1%, interdecay system, absolute time accuracy requires significantly improved experimental determinations of half-lives, but “this cannot occur if we continue to anchor twenty-first century geochronology to often incomplete, 1970s-era nuclear physics reports.” And “what our community appears to lack is the confidence to acquire fundamental new knowledge of decay constants with significantly improved precision and accuracy using the best contemporary facilities.” Perhaps what is needed is for the geochronological community to abandon their assumption of constant radioisotope decay rates!
Limitations of age comparisons
Determining the 238U and 235U decay constants and half-lives from age comparisons has been attempted recently by several geochronologists (Mattinson 2000, 2010; Schoene et al. 2006). However, as Begemann et al. (2001) rightly point out, changing perspectives on which criteria guarantee that a geological event is truly “point-like” in time may (sooner or later) force a revision of some of the results based on this approach. At present, more often than not, the uncertainties assigned to decay constants and half-lives so derived just reflect the reproducibility, not the accuracy. In those experiments the Pb-Pb ratios of zircon and other mineral grains were obtained and compared with the U-Pb ratios obtained by the same mass spectrometry analyses, as per equation (13) above. To legitimately do so there are requirements that need absolutely to be met:
- The Pb-Pb ratios have to be interpreted as to what components of them are primordial (that is, the initial or inherited Pb), what components are due to U decay, and what components have been lost due to leakage. In other words, the 207Pb/206Pb, 238U/206Pb, and 235U/207Pb have to be concordant, which rarely is the case. Otherwise, there is a high risk of obtaining incorrect comparisons due to variable common Pb or open-system processes (Tera and Carlson 1999).
- The uncertainties in the determined radiogenic Pb-Pb ratios (that is, the Pb interpreted to have been derived by U decay) have to be propagated into the derived decay constants and half-lives.
- The uncertainties now known to be in the 238U/235U ratios must be propagated into the derived decay constants and half-lives.
Ironically, charged by the IUGS with the responsibility of providing definitive resolution and guidance on determining from critical review and evaluation of all previous measurements and evaluations of the 238U and 235U half-life values, Villa et al. (2016) did not substantiate or even reiterate the call by Schön, Winkler, and Kutschera (2004) for new direct counting determinations of the 238U and 235U half-lives with at least comparable, if not improved, accuracy. The fact that they do not call for such new experiments indicates a recognition that besides the requirements stated by Begemann et al. (2001) and Schön, Winkler, and Kutschera (2004), there are difficulties in doing more accurate determinations of the 238U and 235U half-lives that make performing such new direct counting experiments somewhat futile.
Difficulties in accurate experimental determinations of the 238U and 235U half-lives
Among these difficulties is the choice of counting instrument. This appears to have escaped the notice of the experimenters and geochronologists. In Fig. 9 it is clearly evident that apart from the 238U half-life determination in 1935, all the experiments prior to 1960 yielded values significantly and distinctively higher that those obtained in the 1960 and 1971 determinations. One potential reason is immediately obvious from the details provided in Table 2. The pre-1960 experiments were all done using ion chambers to count the α-activity specific to 238U, whereas the 1960 and 1971 experiments used a liquid scintillator and a proportional counter respectively. Any new experiments would ideally therefore need to use several different types of counting instruments with the same counting geometry and the same samples to check whether the results obtained are biased by the choice of counting instrument type.
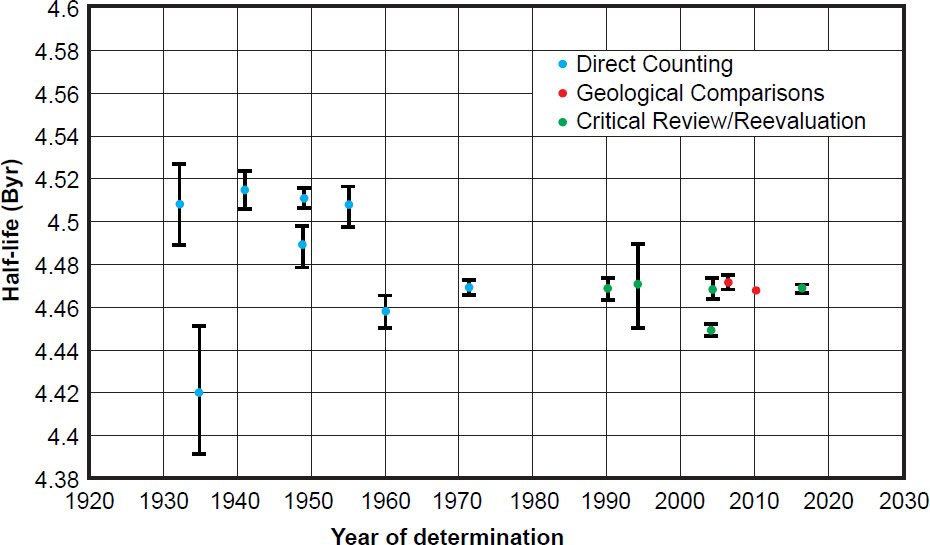
Figure 9. Plot of each 238U half-life determination versus the year of its determination, color-coded according to the method of its determination. The error bars for each determination are also plotted from the error values listed in Table 2.
| Year | Half-Life (Byr) | Uncertainty (Byr) | Instrument | Notes | Source |
|---|---|---|---|---|---|
| 1932 | 4.508 | ±0.018 | Ion chamber | Grid collimator | Kovarik and Adams (1932) |
| 1935 | 4.42 | ±0.03 | Ion chamber | Intermediate geometry | Schiedt (1935) |
| 1941 | 4.514 | ±0.009 | Ion chamber | 2π geometry | Curtiss, Stockman, and Brown (1941) |
| 1949 | 4.511 | ±0.005 | Ion chamber | 2π geometry, Natural U | Kienberger (1949) |
| 1949 | 4.489 | ±0.010 | Ion chamber | 2π geometry, 238U | Kienberger (1949) |
| 1955 | 4.507 | ±0.009 | Ion chamber | Kovarik and Adams (1955) | |
| 1957 | 4.56 | ±0.03 | Ion chamber | 2π geometry | Leachman and Schmitt (1957) |
| 1960 | 4.457 | ±0.007 | Liquid scintillator | 4π geometry | Steyn and Strelow (1960) |
| 1971 | 4.4683 | ±0.0024 | Proportional counter | Intermediate geometry | Jaffey et al. (1971) |
| 1990 | 4.468 | ±0.005 | Reevaluation of non-neutron nuclear data | Coursol, Lagoutine, and Duchemin (1990) | |
| 1994 | 4.47 | ±0.020 | Reevaluation of measurement data | Duchemin, Coursol, and Bé (1994) | |
| 2004 | 4.468 | ±0.005 | Weighted mean | Critical review of corrected experimental data | Schön, Winkler, and Kutschera (2004) |
| 2004 | 4.449 | ±0.017 | Mean | Critical review of corrected experimental data | Schön, Winkler, and Kutschera (2004) |
| 2006 | 4.4712 | ±0.0031 | Geological comparisons | Schoene et al. (2006) | |
| 2010 | 4.4674 | Geological comparisons | Mattinson (2010) | ||
| 2016 | 4.4683 | ±0.0096 | Critical review of experimental data | Villa et al. (2016) |
In contrast, there are no discernible differences in the determined 235U half-life values using different types of counting instruments. In their critical review and reevaluation Schön, Winkler, and Kutschera (2004) discounted the Kienberger (1949) ion chamber determination because it was so obviously in their view an outlier with huge uncertainties, as can be clearly seen in Fig. 10. Yet Schön, Winkler, and Kutschera (2004) did not state any other specific reason, just discounting only the Kienberger (1949) 235U half-life determination of 880 Myr (not his 238U half-life determination using the same equipment) because the result was “inconsistent.” Maybe Kienberger (1949) had a better grasp of his experimental error sources than later experimenters did. If the Kienberger (1949) 238U half-life determination using the same equipment is acceptable, the rejection of his 235U half-life determination may only be due to the assumption of constant decay rates made by Schön, Winkler, and Kutschera (2004). Otherwise, the 235U half-life values determined by ion chambers vary from 684 Myr to 753 Myr, while the 235U half-life values determined by solid-state detectors vary from 685 Myr to 713 Myr (table 3). The latter is definitely a narrower range better centred over the accepted Jaffey et al. (1971) value determined by a proportional counter. And whereas Schön, Winkler, and Kutschera (2004) suggested future experiments might harness liquid scintillators as an appropriate counting instrument, they discounted the Bueno and Santos (1993) experiment which used both a gas scintillator and a NaI (Tl) scintillator for α-γ coincidence determinations, but that was due more to their use of a correction factor being deemed inappropriate.
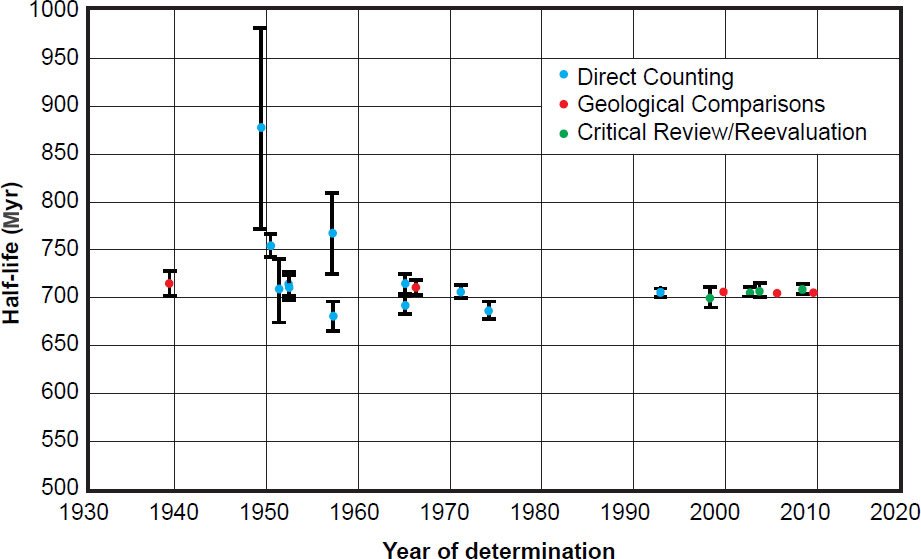
Figure 10. Plot of each 235U half-life determination versus the year of its determination, color-coded according to the method of its determination. The error bars for each determination are also plotted from the error values listed in Table 3.
| Year | Half-Life (Myr) | Uncertainty (Myr) | Instrument | Notes | Source |
|---|---|---|---|---|---|
| 1939 | 713 | ±16 | Mass spectrometer | Geological comparison using activity ratios of U ores of “known” ages | Nier (1939) |
| 1949 | 880 | ±110 | Ion chamber | Specific activity by subtracting ion-chamber 238U specific activity | Kienberger (1949) |
| 1950 | 753 | ±22 | Ion chamber | Specific activity of 235U | Knight (1950) |
| 1951 | 707 | ±33 | Ion chamber | Measured from activity ratio | Sayag (1951) |
| 1952 | 713 | ±16 | Ion chamber | Specific activity of 235U | Fleming, Ghiorso, and Cunningham (1952) |
| 1952 | 710 | ±16 | Correction of Knight (1950) | Fleming, Ghiorso, and Cunningham (1952) | |
| 1957 | 767 | ±43 | Ion chamber | Measured from activity ratio | Clark, Spencer-Palmer, and Woodward (1957) |
| 1957 | 684 | ±15 | Ion chamber | Measured from activity ratio | Würger, Meyer, and Huber (1957) |
| 1965 | 692 | ±9 | Si solid state detector | Measured from activity ratio | Deruytter, Schroder, and Moore (1965) |
| 1965 | 713 | ±9 | Solid state detector | Specific activity of 235U | White, Wall, and Pontet (1965) |
| 1966 | 708.6 | ±7.3/2.9 | Mass spectrometer | Geological comparison using activity ratios of U-bearing minerals of “known” ages | Banks and Silver (1966) |
| 1971 | 703.81 | ±0.48 | Proportional counter | Specific activity of 235U | Jaffey et al. (1971) |
| 1974 | 685 | ±9 | Si solid state detector | 235U activity using central state branching ratio | Deruytter and Wegener-Penning (1974) |
| 1993 | 704 | ±1 | Gas and NaI scintillator | Specific activity of 235U by α-γ conincidence | Bueno and Santos (1993) |
| 2000 | 703.05 | Geological comparisons of zircons <200 Ma | Mattinson (2000) | ||
| 2003 | 703.7 | ±1.1 | Critical review—International Union of Pure and Applied Chemistry report | De Laeter et al. (2003) | |
| 2004 | 706 | ±7 | Mean | Critical review of corrected experimental data | Schön, Winkler, and Kutschera (2004) |
| 2004 | 704 | ±1 | Weighted mean | Critical review of corrected experimental data | Schön, Winkler, and Kutschera (2004) |
| 2006 | 703.06 | ±0.04 | Geological comparisons (anchored to 238U half-life) | Schoene et al. (2006) | |
| 2009 | 706 | ±9 | Mean | Reevaluation of corrected (Schön, Winkler, and Kutschera 2004) 235U decay data | Xiaolong and Baosong (2009) |
| 2009 | 704 | ±1 | Weighted mean | Reevaluation of corrected (Schön, Winkler, and Kutschera 2004) 235U decay data | Xiaolong and Baosong (2009) |
| 2010 | 703.05 | ±0.58 | Geological comparisons (anchored to 238U half-life) | Mattinson (2010) |
In any case, no one thus far has attempted to explain why there is this fundamental difference in the 238U half-live values obtained using ion chambers versus using a liquid scintillator or a proportional counter. It could be due to the media used in each instrument through which the α-particles have to travel from the sample to the detector. In the early ion chambers this was typically ambient air, whereas in the proportional counter used by Jaffey et al. (1971) they used a gas mixture of argon with 10% methane at low pressure, which is now typically used and is colloquially referred to as P10. In contrast, in the liquid scintillator used by Steyn and Strelow (1960) the medium would have been a solvent consisting of aromatic organics such as benzene or toluene, with typically some form of a surfactant, and small amounts of other additives known as “fluors” or scintillators. The choice of medium used within the instrument thus determines how the α-particles will interact with it. A medium with larger molecules will more likely scatter some α-particles so they are not counted, as will a denser medium in which the molecules are closer together. Thus any new experiments would ideally need to compare the effects of different media in different counting instruments. Establishment of accurate quench curves for each type of solvent is critical in determining the counting efficiency. One wonders why they don’t attempt these experiments in a vacuum, as that would eliminate the problem of the medium.
Allied to the choice of instrument type is the geometry of the sample sizes to the sizes of the detector windows, which is related to the construction and configuration of the instruments. For example, in the proportional counter used by Jaffey et al. (1971) shown in Fig. 3 the geometry angle is from the edges of the sample H to the edges of the detector aperture K. It should be immediately evident that any α-particles which have emission trajectories outside that angle, for example, are “shot” sideways, will merely hit the walls J of the instrument and not be counted. That is why it was argued that the instruments with the widest counting angles should have given the best results, because the detectors count more and miss fewer α-particles. This is why counting geometry corrections are so fundamentally important to accurate measurements. However, if the direction of α-particles emitted is random, then one can treat the detector as receiving a random sample within a given solid counting angle. All of the emitted α-particles can be calculated using the ratio of the solid angle of the detector with the solid angle of a sphere surrounding the α-source. However, that factor, and whether natural or enriched uranium was used as the samples to be counted, does not appear to be more important than the type of instrument used.
The next major factor affecting the accuracy of the 238U half-life determinations is the fact that the α-particles emitted by 238U decay span a wide range of energies. Most of the α-particles emitted by 238U decay have a narrow band of energies that when count numbers are plotted against energies results in a distinctive and large 238U peak (see fig. 6 again). However, some α-particles emitted by 238U decay have lower and higher energies on the edges of that peak and even beyond the peak’s tails. Added to that is the problem that the energies of some of the α-particles emitted by 235U decay overlap with not just the tails of the energies of the 238U peak, but also with the energies within the 238U peak. And there will always be background α-particles across the full energy spectrum. Of course, this was the main reason why several experimenters chose to use separate samples enriched in 238U and 235U respectively, so as to minimize and even eliminate the overlapping of the energies of α-particles from 238U and 235U (and 234U). Nevertheless, there still remain two issues crucial to the accurate determination of the 238U half-life and the quoted uncertainties. First is the energy bandwidth chosen for measuring the number of counts (α-particles) in the 238U peak. And second, how many counts across the remainder of the energy spectrum should be attributed to background α-particles, or other ionizing radiation. Both of these issues will apply to all determinations of the 238U half-life regardless of the counting instrument used and its counting geometry, and whether natural or enriched uranium is used for the counting sample.
But the type of sample does introduce another factor. For natural uranium samples, how much of the total α-activity measured is due to 238U decay? In the experiments already done, the contribution of 238U was taken as 48.875%, assuming ≈2.25% of the α-activity was due to 235U, secular equilibrium between 238U and 234U, and a virtually constant 238U/235U ratio. Where it was assumed 238U and 234U were not in secular equilibrium, the α-activity due to 238U was taken to be 49.4 ± 0.5% of the total U α-activity. However, the issue here is these are assumptions. To minimize the extent of any unrealized variations in these assumptions so that they can be factored into the determined 238U half-life values, ideally in each experiment the natural uranium samples used should be checked for the secular equilibrium between 238U and 234U, and the 238U/235U ratio. So this is why enriched samples are preferred, but even then such samples need to be carefully analyzed not only to determine their purity, but also the identity of any impurities so that their effects can be factored into the 238U half-life determination. Jaffey et al. (1971) used two batches of 238U with very high isotopic enrichments, namely, 99.9790 ± 0.0001 atom % and 99.9997 ± 0.0001 atom %, but some atoms of 235U and 234U were still present which would still have had some minor effects on the statistics of the 238U half-life determination.
Similar considerations apply to the 235U half-life determinations. Two procedures have been adopted—measuring the specific activity of 235U primarily using samples enriched in 235U, or measuring the 238U/235U activity ratio using natural uranium samples (table 3). However, both procedures in effect rely on being able to measure the total energy spectrum of α-particles and to then distinguish and quantify accurately the separate α-particle spectra contributions of 238U, 235U, and 234U. The 235U α-spectrum in particular is difficult to distinguish and separate from the total α-spectrum for natural uranium samples because it consists of a central peak and two bordering minor peaks either side of it, but those two bordering minor peaks are hidden under the large 238U and 234U peaks. This issue has already been described and discussed in detail above and by Schön, Winkler, and Kutschera (2004). Then similar to determining the 238U peak, there are the questions of the energy bandwidth chosen for measuring the number of counts (α-particles) in the 235U peak, and how many counts across the remainder of the energy spectrum should be attributed to background α-particles. Allied to all these considerations is the necessary use of the 238U/235U ratio in these two procedures, and also when a mass spectrometer is used to analyze for the isotopic ratios. Up until recently the 238U/235U ratio has been assumed to be essentially constant, though it is now known to vary significantly from rock to rock and mineral to mineral (see below). All these considerations have resulted in much uncertainty about the accuracy of the 235U half-life determinations, and very much larger error margins being attributed to them (0.07–5.6%) compared with the 238U half-life determinations (0.05–0.68%), as calculated from the quoted error margins listed in Tables 2 and 3.
Finally, there are issues related to the counting efficiency and statistics, such as the self-shielding of finite-thickness solid samples, the low specific α-activities, the detection of very low-energy α-decays, and problems with detector efficiencies. Because the uranium samples used are solid and thus densely packed grains containing large U atoms compared to the size of the α-particle, some α-particles will inevitably be absorbed within the samples, never getting emitted beyond the samples’ surfaces, and thus are not counted. Then there are the low levels of specific α-activities, even for highly enriched samples. Thus one has to be sure that the counting period is long enough to accumulate a large enough count of the specific α-particles to ensure good counting statistics of the peaks compared to the background. The same applies to the detection and counting of the very low-energy α-particles. And the last important aspect to be considered are problems with detector efficiencies. The list of those includes, but is not restricted to, the reaction and recovery times of the ionizing gas in the ion chamber or proportional counter, and of the scintillator in solution in the liquid scintillator, and the dead-times in between the generated pulses being transmitted to the recording end of the instrument and pulse pileups during high counting episodes, that is, through its electronics.
Each of these issues add a measure of uncertainty to the determined 238U and 235U half-life values and the error margins that should be reported, so that the accumulative impact of them all is substantial. Furthermore, the uncertainties due to each of these issues makes it very difficult to be absolutely certain that the half-life values have been determined accurately and to also quantify what the total uncertainty might be for each determined half-life value. The combined effects of these uncertainties have a huge impact on the accuracy and uncertainties of the U-Pb and Pb-Pb ages calculated using the determined 238U and 235U half-life values.
Geological comparisons
The second approach used by secular scientists to determine the 238U and 235U decay half-lives has been to use geological samples (uranium ores or uraniumbearing minerals from various rock units) whose ages have been measured by the U-Pb methods to obtain their 206Pb/207Pb ratios (Dickin 2005; Faure and Mensing 2005). Those measured 206Pb/207Pb ratios are then used to evaluate the 238U and 235U half-lives via the present-day 235U/238U activity ratio. In some instances, the ages of the samples were crosschecked with the Ar-Ar ages of their minerals. These procedures all essentially involve circular reasoning, because it is being assumed the radioisotope dating methods, principally the U-Pb method, give the reliable dates from which the 238U and 235U half-lives can be calibrated to bring the U-Pb and other radioisotope ages into agreement. It should be noted, however, that this is hardly objective, because all the radioisotope ages of rocks could be wrong due to the underlying unprovable and suspect assumptions on which all the radioisotope dating methods are based.
Nier (1939) was the first to attempt to determine the 238U and 235U half-lives by using uranium ores whose ages had been determined by measuring 206Pb/238U and 207Pb/235U ratios. He then used the measured 206Pb/207Pb ratios as a function of age to evaluate the present-day ratio R = (activity of 235U)/ (activity of 238U), and finding it to have a value of 0.046 ± 0.001, from it calculated the 235U half-life. Banks and Silver (1966) repeated the same procedure on uranium-containing minerals that were relatively young so as to minimize any effect of partial leaching out of the Pb component. Again the 206Pb/207Pb ratio was used as being equivalent to the activity ratio R. So assuming the known 238U half-life, the 235U half-life could be calculated.
More recently, Mattinson (2000, 2010) and Schoene et al. (2006) attempted to reassess and refine the determined values of the 238U and 235U half-lives by obtaining multiple analyses of zircon and xenotime grains from various rock units for their 238U/206Pb, 235U/207Pb, and 207Pb/206Pb ages. Schoene et al. (2006) used 11 rock samples ranging in age from 0.1 to 3.3 Ga, and obtained large statistically equivalent datasets yielding 207Pb/206Pb dates that were systematically older than the 206Pb/238U dates by ~0.15% in Precambrian samples to as much as ~3.3% in Mesozoic samples. They concluded that these results suggested inaccuracies in the mean values for one or both of the 238U and 235U half-lives, so they used these datasets to calculate a new ratio of the 238U and 235U half-lives with very high precision (± ~0.02%) and thus new values for the 238U and 235U half-lives. In contrast, Mattinson (2000) used only zircon grains from < 200 Ma rock samples, while Mattinson (2010) restricted his study to zircon grains from 14 rock samples in the age range 400–560 Ma. Such a restriction in itself already introduced a bias. However, in both studies it was argued that these younger samples were chosen so that the corrections for intermediate daughter isotope disequilibrium would be low. Mattinson (2010) also selected the zircon grains for the apparent perfect concordance of their 238U/206Pb, 235U/207Pb, and 207Pb/206Pb ages except for small deviations that could be explained by errors in the 238U and 235U half-lives used in the age calculations. In all these studies the 235U half-life was determined from the 235U/238U activity ratio by assuming the 238U half-life had first been accurately determined, and by assuming the isotopic ratio 238U/235U is constant at 137.88.
This method has the disadvantage that it involves geological uncertainties, such as whether all isotopic systems closed at the same time in the various minerals in the same rocks and remained closed. However, it is claimed to still provide a crucial check on the laboratory determinations by direct physical counting. Nevertheless, this approach entails repeated multi-chronometric U-Pb dating of minerals from the same rocks and cross-calibration of three different dates obtained from the radioisotopic U-Pb age system by adjusting the 235U half-life and thus the 235U/207Pb age so as to force agreement with the 238U/206Pb age assuming the 238U half-life is the accurately determined standard and the isotopic ratio 238U/235U is a constant (Begemann et al. 2001). Thus, because the half-life of 238U is claimed to be the most accurately known of all relevant radionuclides, this usually amounts to expressing ages in units of the half-life of 238U.
Critical review and reevaluation
Several studies focused instead on critically reviewing and reevaluating all the laboratory direct counting determinations of the 238U and 235U half-lives. For example, Coursol, Lagoutine, and Duchemin (1990) and Duchemin, Coursol, and Bé (1994) used the γ-ray energies and emission probabilities of each of the isotopes in the 238U decay chain, and the α- and β-particle emission probabilities to reevaluate the 238U decay half-life value. Similarly, de Laeter et al. (2003, 787) provided a brief report on their assessment of the recommended values for the 238U and 235U half-lives, presumably based on a review of the then extant literature.
The most comprehensive and thorough critical review of the experimental data for the half-lives of 238U and 235U is still that of Schön, Winkler, and Kutschera (2004). They compiled all the experimental direct counting data and divided them into four groups.
In Group 1 were the direct measurements of the specific activity of 238U and 235U (decay rate per microgram of the particular isotope). They listed the requirements to thus accurately determine the 238U and 235U half-lives as:
- accurate chemical analysis of the sample material for the amount of uranium present;
- mass-spectrometric analysis to determine the fraction of uranium present as the desired isotope;
- a sample-preparation technique that ensures each sample to be decay-counted contains an accurately known sample mass. Then an accurate and precise measurement of the α-emission rates of 238U and 235U requires:
- measurement of the total α-emission rate by employing a counter with well-known efficiency, and achieving a sufficient number of counts to keep the statistical counting uncertainty small;
- analysis of the α-energy spectrum using the same or another detector with sufficient energy resolution to derive the fraction of the total α-activity associated with the 238U and 235U isotopes (peak selection).
Counting statistics are Poisson statistics, so the uncertainty is the square root of the number counted. Since uranium today has a very long half-life, it would take a very long time to get enough counts for a small uncertainty.
Additional requirements they cited are the accurate determinations of the counting solid angle (if not 4π), and scattering effects due to the sample backing, sample layer, counter walls, filling gas, and apertures must also be reliably estimated. Furthermore, a high degree of enrichment of the 238U and 235U isotopes in the respective samples used also minimizes systematic uncertainties, in particular with respect to the α-spectrum evaluation (see fig. 6 again). Thus they highlighted that compared to other experimental determinations in the literature, the measurements by Jaffey et al. (1971) were characterized by sample materials with very high isotopic enrichments—(99.9790 ± 0.0001) atom % and (99.9997 ± 0.0001) atom % for the two batches of 238U, and (99.99886 ± 0.00002) atom % for 235U.
In their Group 2 Schön, Winkler, and Kutschera (2004) placed the “semi-direct” measurements of 238U employing natural uranium samples. With this method the total α-activity of natural uranium was measured and the contribution of 238U was taken as 48.875%, assuming ≈2.25% of the α-activity was due to 235U, secular equilibrium between 238U and 234U, and a virtually constant 238U/235U ratio. Where it was assumed 238U and 234U were not in secular equilibrium, the α-activity due to 238U was taken to be 49.4 ± 0.5 % of the total U α-activity.
Then in Group 3 they placed measurements of 235U relative to 238U. This was done by using an α-particle spectroscope to determine the activity ratio
The α-energy spectrum exhibits three groups of peaks attributed to 238U, 235U, and 234U (see fig. 6 again). But only ≈85% of the 235U α-activity is recorded in its clearly visible central peak between the dominant 238U and 234U peaks. However, the rest of the 235U α-spectrum is hidden under the 238U and 234U peaks. So as already indicated above, there are problems inherent in estimating the intensity of the central peak when there are associated with it large relative uncertainties in the estimated branching ratio, which Schön, Winkler, and Kutschera (2004) estimated is 85.6 ± 1.3 % as the average of measurements by Ghiorso (1951), White, Wall, and Pontet (1965), Jaffey et al. (1971), and Deruytter and Wegener-Penning (1974). Also, the number density ratio N(235U)/N(238U) (usually written as 235U/238U) was assumed to be constant.
Finally, Schön, Winkler, and Kutschera (2004) placed in their Group 4 the indirect determinations of 235U relative to 238U. These involved the determination of the present activity ratio R [equation (12) above] by using inverse geochronological methods from the U-Pb ages of a range of uranium-bearing minerals measured by a mass spectrometer (Banks and Silver 1966; Nier 1939). From the total 207Pb/206Pb ratio the radiogenic 207Pb*/206Pb* ratio was derived by subtracting the initial (207Pb/206Pb)i ratio at the time of mineral formation. The basic transcendent relationship then is
The present activity ratio R is thus determined from the ages of the minerals (t), the half-life ratio is obtained from the present 235U/238U ratio (which is assumed to be constant), and thus the 235U half-life is determined relative to the 238U half-life (which is assumed to have been accurately determined). Equation (13) can be represented by a group of curves in a plot of 207Pb*/206Pb* versus t for a given λ238, each curve characterized by a certain activity ratio R. The best fit to the data determines the activity ratio R.
Having critically evaluated each of the determinations of the 238U and 235U half-lives in these four groups, Schön, Winkler, and Kutschera (2004) produced tables of their revised half-life values. Some half-life determinations were excluded from their further consideration due to perceived lack of experimental details to assess their validity, inconsistencies, or methodological error. However, this is not usually the way the scientific method works. Instead, exclusion of data must be performed by the experimenters themselves when they become aware of a demonstrable problem in any of their measurements. Nevertheless, when Schön, Winkler, and Kutschera (2004) then calculated the averages from the remaining accepted revised half-life values, the internal uncertainty (from “error propagation” and by taking into account possible correlations between the data) and the external standard uncertainty (based on the scatter of the data) were estimated, and the larger of the two was usually used (Winkler 1998). Their resultant mean and weighted mean values of the 238U and 235 half-lives and their uncertainties were dominated by the results of Jaffey et al. (1971) for both isotopes, and therefore essentially reproduced them. However, they noted that for 238U measurements carried out with natural uranium only the revised values and associated uncertainties assuming that 238U and 234U are not in equilibrium were used in their final averaging calculations. And the results obtained for the 235U half-life relative to the 238U half-life depend on the adopted value of the latter, that is, the Jaffey et al. (1971) value.
Xiaolong and Baosong (2009) evaluated the published 235U decay data for γ-ray, electron, and α-particle emission probabilities and produced a revised 235U decay scheme. They also tabulated the original experimentally-determined 235U half-life values and the revised values of Schön, Winkler, and Kutschera (2004), but then merely recalculated virtually the same mean and weighted mean values.
Finally, Villa et al. (2016) were tasked by the International Union of Geological Sciences (IUGS) with the independent reanalysis and reevaluation of the available data on determinations of the 238U and 235U half-lives. Yet they merely reiterated the 238U half-life value recommended by Schön, Winkler, and Kutschera (2004), the same as the Jaffey et al. (1971) value, though they noted the discussions of uncertainties by Mattinson (1987, 2010) and Schön, Winkler, and Kutschera (2004). Then after a lengthy discussion of the dependence of the determination of the 235U half-life value on the adopted 238U half-life value, the effect of the very recently recognized variability in the 238U/235U ratio, and various analytical effects such as instrumental mass fractionation, Villa et al. (2016) saw no reason to modify the Jaffey et al. (1971) value for the 235U half-life and its associated measurement uncertainty as amended by Schön, Winkler, and Kutschera (2004).
Results of the 238U and 235U Decay Half-Life Determinations
During the last 84 years, numerous determinations of the decay constants and half-lives of 238U and 235U have been made using these three methods. The results are listed with details in Tables 2 and 3. The year of the determination versus the value of the 238U and 235U decay half-lives are plotted in Fig. 9 and Fig. 10 respectively, along with the error bars for each determination as per the error values or uncertainties listed in Tables 2 and 3 respectively. In each case the data points plotted have been color-coded the same to differentiate the values as determined by the three approaches that have been used—direct counting, geological comparisons using the U-Pb methods, and critical review and re-evaluation.
Discussion
The decay of 238U and 235U to 206Pb and 207Pb, respectively, forms the basis for one of the oldest methods of geochronology (Dickin 2005; Faure and Mensing 2005). While the earliest studies focused on uraninite (an uncommon mineral in igneous rocks), there has been intensive and continuous effort over the past five decades in U-Pb dating of morecommonly occurring trace minerals. Zircon (ZrSiO4) in particular has been the focus of thousands of geochronological studies, because of its ubiquity in felsic igneous rocks and its claimed extreme resistance to isotopic resetting (Begemann et al. 2001). Such resistance to isotopic resetting is claimed because it engenders confidence in using zircon U-Pb geochronology. Yet there are documented experiments and many measurements on natural zircons that demonstrate mobility of U and Pb in zircons and thus isotopic resetting due to radiation damage and diffusion (for example, Cherniak and Watson 2003; Ewing et al. 2003; Seydoux-Guillaume et al. 2015; White and Ireland 2012).
No decay constant or half-life of any radionuclide used for geochronology has been (or, arguably, can be) more-precisely measured than those of 238U and 235U—a consequence of the mode of decay (alpha), favorably short half-lives, and the availability of large quantities of isotopically pure parent nuclides (Begemann et al. 2001). However, this assertion is hard to accept when the most recent experimental measurements accepted as reliable by the geochronology community (Schön, Winkler, and Kutschera 2004; Villa et al. 2016) are those made by Jaffey et al. (1971), now more than 45 years ago. At the time Jaffey et al. (1971) quoted precisions (recalculated to 95%-confidence limits) of 0.11% for 238U and 0.14% for 235U, with the somewhat cryptic statement that “systematic errors, if present, will no more than double the quoted errors.” Thus it is hard to believe those quoted errors, given the measurement apparatus used in these determinations. Nevertheless, when Schön, Winkler, and Kutschera (2004) recommended the continued adoption of the Jaffey et al. (1971) values for 238U and 235U decay half-lives they essentially doubled the uncertainties.
Not one U-Pb dating method
Recognizing both the high precision of the half-life measurements and the robustness of U-Pb geochronology on such minerals as zircon, in 1976 the IUGS Subcommission on Geochronology recommended that “The U decay constants by Jaffey et al. (1971) must be the basis for any standard set of decay constants” (Steiger and Jäger 1977). A question not addressed by this recommendation, however, is the complication that there are essentially five distinct methods for calculating a “U-Pb” age from a given set of U-Pb isotope data, each of which involves the effects of the uranium decay constants or half-lives in different ways (Begemann et al. 2001). These are, with the asterisks indicating the radiogenic portions of the respective isotopes:
- The 206Pb/238U age, from t = ln (1 + 206Pb*/238U)/λ238 [equivalent to equation (6) above]
- The 207Pb/235U age, from t = ln (1 + 207Pb*/235U)/λ235 [equivalent to equation (7) above]
- The 207Pb/206Pb age, from 207Pb*/206Pb* = [(eλ235t –1)/(eλ238t –1)] × (235U/238U)today [equivalent to equations (9) and (10) above]
- The upper or lower concordia-intercept age, from the intercepts of the linear trend of the analyses of several cogenetic samples with the concordia curve on one of the U-Pb concordia diagrams—either “conventional,” with the horizontal axis x = 207Pb*/235U, and the vertical axis y = 206Pb*/238U (Wetherill 1956), or “Tera-Wasserburg” (Tera and Wasserburg 1972) with x = 238U/206Pb*, y = 207Pb*/206Pb*.
- The “concordia age” (Ludwig 1998), which finds the most probable age for a U-Pb analysis whose 206Pb/238U-207Pb/235U age-concordance can be assumed.
Each of these methods involves the two U decay constants or half-lives in different ways (Ludwig 1998, 2000; Mattinson 1994a, b), so that there is really no single “U-Pb age,” and therefore no clear-cut way to implement the recommendations of Steiger and Jäger (1977) in this regard. Thus Begemann et al. (2001) concluded that the most straightforward solution is to select the 206Pb/238U system as the standard for age normalization. Ages derived from this system are supposedly the least affected by decay constant or half-life uncertainties of any simple system, and they are without the complications of the dual-system ages of schemes (3) through (5) above, whose precision advantages apparently only occur for Precambrian ages in any case. At any rate, because of the complicated differential effects of the two U half-lives, it is recommended that for U-Pb dates where accuracy is important the quoted uncertainties in “Pb-Pb,” concordia-intercept, or “concordia” ages always include the effect of the half-life uncertainties. Moreover, Begemann et al. (2001) suggested it is possible that experiments on natural minerals can improve the precision of the ratio of the two U half-lives (Mattinson 1994a), in which case further improvement in the accuracy of ages of types (3)–(5) can be realized.
Dependence on the 235U half-life on the 238U half-life
There is no question that every determination of the 235U half-life depends on accurately knowing the 238U half-life. This is clearly evident from the reports of the experiments, and the critical reviews and re-evaluations of Schön, Winkler, and Kutschera (2004), and Villa et al. (2016). Equations (12) and (13) above show how the 238U half-life is used in the calculation of the 235U half-life. This is not only apparent in the direct counting experiments, but it is the essential procedure in all geological comparisons, whether using rock or mineral samples (Villa et al. 2016).
This necessity compounds the problems and uncertainties of accurately determining the 235U half-life. The uncertainties associated with determining the 238U half-life add to the greater uncertainties in determining the 235U half-life, as already discussed. Just the fact that the dominant 238U α-spectrum and peak overlap the very minor 235U α-spectrum and peaks, plus the background and the strong 234U α-spectrum and peak (fig. 6), complicates both the 238U and 235U half-life determinations, especially the latter because of the branching ratio that results in the two secondary peaks bordering the tails of the primary 235U peak (fig. 7). While using separate samples enriched in 238U and 235U respectively for separate determinations, as Jaffey et al. (1971) did, alleviates some of these issues, one still has to assume secular equilibrium between 238U and 234U. 234U is produced by decay of 238U in the sample (ingrowth, see fig. 1) over the time of the experiment, from sample preparation through the counting period. That disequilibrium has been recognized as a problem is noted by Villa et al. (2016), who pointed out that geological comparisons endorsed by Steiger and Jäger (1977) were later considered problematic by Begemann et al. (2001), and then further revealed as a definite problem by the work of Villa (2010). The counting period has to be long enough to gather sufficient counts to keep the statistical counting efficiency small, but that allows for ingrowth of 234U. And then there is always the background in the total α-energy spectrum which has to be accurately evaluated and subtracted from the 238U, 235U, and 234U peaks and bordering tails.
Variations in the critical 238U/235U ratio
There is also no question about the critical necessity of knowing the present day 238U/235U ratio in both determining the 238U and 235U half-lives, and in calculating U-Pb and Pb-Pb ages using the 238U and 235U half-lives. It is also worth noting here, that if there was accelerated decay in the past during some catastrophic event such as the Flood (Vardiman, Snelling, and Chaffin 2005), and the 235U and 238U decay constants changed proportionately, then the 238U/235U ratio may have been higher in the past, say at the time prior to the Flood. The Oklo scientists (see below) think it may have been as high as 7% (instead of today’s 0.72%) 235U at the time of the nuclear reactions (1.8 Ga, that is, Precambrian and therefore pre-Flood), due to the different half-lives of 235U and 238U (Chaffin 2005).
In the critical review and re-evaluation of Schön, Winkler, and Kutschera (2004) reported above, their group 2 238U half-life determinations and their groups 3 and 4 235U half-life determinations all required the use of the 238U/235U ratio, which was assumed to be constant. It appears in equations (12) and (13) as a necessity for calculating the 235U half-life. Likewise, in the essential conditions necessary to achieve reliable 206Pb and 207Pb model ages [equations (6) and (7) above] listed by Faure and Mensing (2005), condition 4 is that the isotopic composition of uranium is normal and has not been modified by isotope fractionation, that is, the 238U/235U ratio is a known constant. Yet if this ratio is a discontinuous function of time then L’Hôpital’s rule cannot be used to determine the initial Pb isotope ratios. Similarly, the 238U/235U ratio appears in equations (9) and (11) for calculating 207Pb-206Pb model ages, is present by implication in U-Pb concordia ages and U-Pb discordia ages (Terra and Wasserburg 1972; Wetherill 1956), and is used to calculate Pb-Pb isochron ages (Faure and Mensing 2005). This demonstrates how the 238U/235U ratio is critically interwoven into the U-Pb dating methods, and thus their viability and accuracy hinge on that ratio being known and being constant.
The first documented variation in the 238U/235U ratio in natural uranium was reported widely in 1976 (Cowan and Adler 1976). However, French scientists had already been investigating this discovery for several years (Baudin et al. 1972; Bodu et al. 1972; Slodzian and Havette 1974). It was in high-grade uranium ore at Oklo in Gabon (Africa) and was well-demonstrated as produced by natural fission having occurred (Cowan 1976; Kuroda 1982; Lancelot, Vitrac, and Allègre 1975). Investigators had been searching for some time for isotopic variations in both uranium ore minerals and uranium ore concentrates (Malyshev et al. 1975), but the precision of the available instruments was inadequate to quantify any likely observed variations (Hamer and Robbins 1960). Nevertheless, the search continued for more than two decades for variations in the abundance of 235U, and thus in the 238U/235U ratio in uranium ores, with not a great deal of success (for example, Apt et al. 1978; Riley and Korsch 1980). But then Richter et al. (1999) reported significant differences in the 238U/235U ratios of uranium ores from six different locations.
Bigeleisen (1996) predicted that uranium isotopes fractionate as a result of nuclear-field shift, which is a consequence of the difference in nuclear sizes and shapes of isotopes. Unless both uranium isotopes are fully ionized (which is not likely) they cannot approach each other close enough for the nuclear field to play any part in their interaction. Yet 235U with an odd number of neutrons has a smaller nucleus relative to nuclei with an even number of neutrons, such as those of 238U. This results in different bond strengths, with 238U preferentially incorporated into the more condensed solid phase. Schauble (2007) confirmed the theoretical calculations and suggested that the nuclear-field shift results in 238U/235U ratios that vary as a function of uranium oxidation state, with the highest 238U/235U ratio in more reduced species. Apparently what Bigeleisen (1996) meant is the electron cloud shifts due to the orientation of the nucleus and thus changes the geometric configuration of the isotope. Moreover, a subsequent ab initio relativistic molecular orbital study for the 238U-235U pairs showed that the nature of ligands surrounding uranium atoms effects isotope fractionation caused by nuclear-shift (Abe et al. 2008, 2010). Thus the mass difference of the various uranium isotopes would be the dominant cause for fractionation if those isotopes are in elemental form.
Only in the last decade though has it been feasible to detect much smaller variations in 238U/235U ratios than in the past. This has been because of the development of multi-collector (MC) ICP-mass spectrometers (MS) that produce a precision of 0.05‰ (per mil) for 238U/235U ratios (Hoffmann et al. 2007), though it easy to be dubious about the quoted error. Thus recent investigations of natural terrestrial and extra-terrestrial samples have revealed variations in the 238U/235U ratios of up to ~5.5‰ and 3.5‰ respectively, that is, parts per thousand (‰ or per mil) deviations from a 238U/235U ratio value of 137.88, the hitherto fore accepted value which has been, and still is, used in all U-Pb age determinations for more than four decades.
This has spawned a concerted effort to find and characterize prepared natural and synthetic uranium samples with certified and thus agreed upon U isotope ratios that can be therefore used as 238U/235U ratio standards against which the deviations in 238U/235U ratios in natural terrestrial and extra-terrestrial samples can be measured (for example, Condon et al. 2010; Richter et al. 2008, 2009, 2010). Thus Richter et al. (2010) reported a study in which a frequently used reference sample NBS960 (or NBL CRM 112a) was reanalyzed in a collaborative effort by several geochemistry laboratories using their MC-ICP-MS and TIMS (thermal ionisation MS) instruments. They found that whereas the consensus 238U/235U ratio value for this standard was previously 137.88, their results indicated that the value is actually 0.031% lower at 137.837 ± 0.015. One wonders though whether they really know the ionization efficiency of their equipment that accurately. Independently, Condon et al. (2010) undertook determinations of the 238U/235U ratios of a suite of three commonly used natural and two synthetic uranium reference materials using a TIMS instrument. They reported the 238U/235U ratio values they obtained for these standards were up to 0.08% lower than the widely used consensus value of 137.88, with the total uncertainty estimated to be <0.02% (2σ) (fig. 11).
Figure 11. Summary plot of the 238U/235U ratio values for natural and synthetic uranium standard samples obtained by Condon et al. (2010), showing the absolute and epsilon ε scales relative to the consensus value of 137.88. The dark grey band reflects the external reproducibility, and the lighter grey band represents the combined standard uncertainty. The black bar represents the certified value and combined uncertainty of standard IRMM 184. All ranges are plotted with a coverage factor of k = 2 (2σ).
Variations in the 238U/235U ratio in uranium ores
Among the natural uranium samples that have been tested for their 238U/235U ratio values are uranium ores and ore concentrates from various types of uranium deposits around the world. Four decades ago Cowan and Adler (1976) reported a statistically significant variation of up to 0.03% in 238U/235U ratio values in uranium ores relative to a “Belgian Congo” ore standard. Interestingly, they found that the variations in the 238U/235U ratio values when plotted grouped into two modes, one for “sandstone” type ores of the Colorado Plateau which were well below the standard value, and one for what they termed “magmatic” type ores whose mode plotted just below the standard value (fig. 12). They suggested the depleted values in the “sandstone” type ores were due to chemical differentiation of the U isotopes during transport of the U in solution in groundwaters and its concentration in the ores. Richter et al. (1999) suggested that the different 238U/235U ratio values for different uranium ores provided unique isotopic “fingerprints” according to their geographic origin. Keegan et al. (2008) confirmed that there were unique U isotopic “fingerprints” for U ore concentrates from three Australian mines, though their 238U/235U ratio values were not significantly different.
Figure 12. Histogram of 235U contents of various global uranium ores as determined by Cowan and Adler (1976) and presented by Bopp et al. (2009). There is a bimodal distribution of samples’ values, with “sandstone-type” ores being offset to lower (235U wt.%) relative to “magmatic-type” ores.
However, Bopp et al. (2009) revisited the data and observations of Cowan and Adler (1976) (fig. 12) and added to them high precision 238U/235U ratio values determined by MC-ICP-MS for six U ore samples representing the same two uranium deposit types. They found that samples from the tabular sandstone-type uranium deposits whose ores precipitated from groundwaters at low temperatures were depleted in 235U, with a total offset of ≈1.0‰ from the hydrothermal uranium deposits whose ores precipitated at high temperatures. They attributed this offset to temperature-dependent fractionation of the U isotopes related to the nuclear-field shift during chemical reduction of U6+ to U4+ in ambient temperature groundwaters during ore deposit formation.
Brennecka et al. (2010a) similarly used MCICP-MS to determine high precision 238U/235U ratio values for a significantly larger suite of forty samples of U ore concentrates from three major uranium depositional settings based on the temperature and redox environment—low-temperature, redox-sensitive sandstone, and black shale deposits; large high-grade, high-temperature, redox-sensitive unconformity-related, vein-type, intrusive, metamorphic core complex and collapse breccia-pipe deposits; and non-redox-sensitive quartz-pebble conglomerate deposits. Their results (fig. 13) show clear evidence that the depositional redox environment in which uranium is precipitated to form the ore deposits is the primary factor affecting 238U/235U fractionation, because the low-temperature deposits are, on average, isotopically ~0.4‰ heavier than uranium deposited at high temperatures or non-redox processes. Furthermore, 238U/235U ratios coupled with 235U/234U ratios in the same samples provide evidence that the redox transition (U6+ to U4+) at low temperatures is the primary mechanism of 238U/235U fractionation, and that preferential leaching from aqueous alteration plays a very limited role, if any, in fractionation of the 238U/235U ratio. They concluded that the direction and magnitude of this fractionation consistently follow the predictions of the nuclearvolume effect associated with heavy elements (Abe et al. 2008, 2010; Bigeleisen 1996; Schauble 2007), and are opposite to the fractionation patterns of lighter elements such as N, S, and Fe.
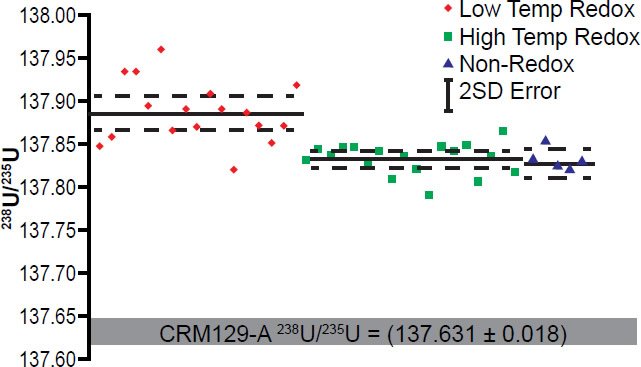
Figure 13. The 40 samples in the Brennecka et al. (2010a) study plotted by depositional style and 238U/235U ratio values. The solid lines represent the average group 238U/235U ratio values and the two standard deviation error is shown for each depositional style in dashed lines. The reported 238U/235U ratio value and external reproducibility of the CRM 129a standard is shown in the black box.
Such investigations also focused on U-bearing minerals, not just on uranium ore deposits (Uvarova et al. 2014), but on U-bearing minerals as accessory minerals in a wide variety of rocks (Hiess et al. 2012). Uvarova et al. (2014) used MC-ICP-MS to determine variations in the 238U/235U ratio values for 126 uranium ore mineral samples (primarily uraninite, but also autunite, fluorapatite, carnotite, brannerite, uranophane, and becquerelite) and nine samples of the rhyolite which is regarded as the source of that autunite mineralization. Represented were ore minerals from calcrete, diagenetic phosphate, quartz-pebble conglomerate, intrusive (magmatic)-related, metasomatic-related, volcanic-hosted, sandstone-hosted, vein-type, and unconformity-related uranium deposits. Their results are plotted in Fig. 14. Mean δ238U values of these uranium minerals relative to NBL CRM 112a were 0.02‰ for metasomatic deposits, 0.16‰ for intrusive, 0.180/00 for calcrete, 0.18‰ for volcanic, 0.29‰ for quartz-pebble conglomerate, 0.29‰ for sandstone-hosted, 0.44‰ for unconformity-type, and 0.56‰ for vein, with a total range in δ238U values from −0.30‰ to 1.52‰.
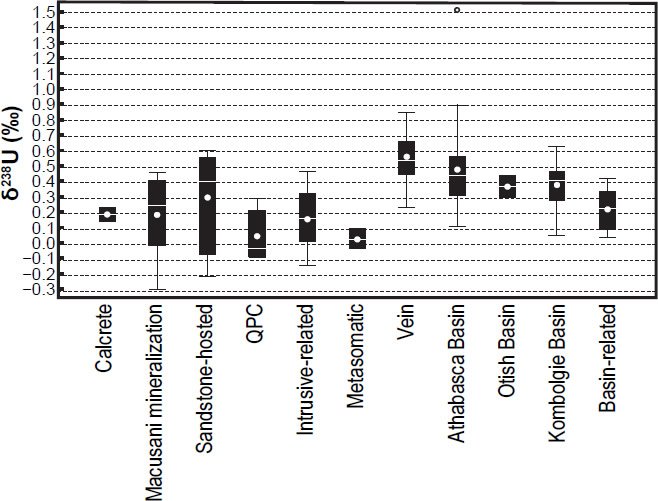
Figure 14. Tukey box-and-whisker plots of δ238U values for uranium ore minerals from various deposits and prospects (after Uvarova et al. 2014). Each box is defined by the 25th and 75th percentiles of the data and represents the inter-quartile range (IQR). The median is represented by a white horizontal line and the mean by the white dot. Whiskers are extended from the box to the last value as <1.5 IQR, towards the maximum and the minimum. Circles beyond the whiskers are considered outliers or anomalies (circles).
Hiess et al. (2012) found that uranium mineralization associated with igneous systems, including low-temperature calcretes that are sources from U-rich minerals in igneous systems, have low δ238U values of ~0.1‰, near those of their igneous sources and thus reflecting the isotopic signature of the crustal source of uranium and the effectiveness of the trapping mechanism. Uranium minerals in basin-hosted deposits have higher and more variable values. High-grade unconformity-related deposits have δ238U values around 0.2‰, whereas lower grade unconformity-type, vein-type, and sandstone-hosted deposits have higher δ238U values of ~0.4‰. They concluded that the 238U/235U fractionation occurs as a function of nuclear-field shift. However, variations in 238U/235U ratios are small in high-temperature magmatic- and metasomatic-related deposits and in cases where mobilized uranium from an igneous protolith is effectively precipitated in a closed system. Significant variations in 238U/235U ratios also occur during Rayleigh fractionation as a result of 238U enrichment as U6+ in the reduced species (mostly uraninite UO2), or during fluid alteration/recrystallization of uraninite wherein 235U is preferentially mobilized into the fluid as the oxidized mobile uranyl (UO22+) ion and its complexes. Therefore, the δ238U values of uranium minerals are controlled by the isotopic signature of the uranium source, the efficiency of uranium reduction in the case of UO2 systems, and the degree to which uranium was previously removed from the fluid, with less influence from temperature of ore formation and later alteration of the ore. All these conclusions have subsequently been further validated by Murphy et al. (2014).
Variations in the 238U/235U ratio in U-bearing accessory minerals
In contrast, Hiess et al. (2012) reported 141 238U/235U determinations on a suite of 58 samples of U-bearing accessory minerals that are used for U-Pb geochronology (zircon, monazite, apatite, titanite, uraninite, xenotime, and baddeleyite), spanning from the Eoarchean (3880 Ma) to the Quaternary (0.24 Ma) and covering a diverse range of igneous and metamorphic petrogenetic settings and geographic locations. Their data set exhibits a range in 238U/235U ratio values of > 5.4‰ (fig. 15), with no clear relation to any petrogenetic, secular, or regional trends. The lowest measured value was 137.743 for monazite derived from the Moacyr pegmatite in Brazil (504.3 Ma), and the highest was 138.490 for titanite from the Fish Canyon Tuff in Colorado (28.4 Ma). The latter titanite and two other samples are not shown in Fig. 15 because of also yielding high 238U/235U values, 138.068 for a titanite metamorphic megacryst from Bear Lake in Canada (1047 Ma), and 138.283 for zircon (likely mantle-derived) from Table Cape in Tasmania, Australia (19.9 Ma). Thus they suggested that variation in 238U/235U between comagmatic minerals (for example, zircon with 137.831 and titanite with 138.490 in the Fish Canyon Tuff, a fractionation of 4.78‰) indicates crystal-chemical control (magmatic or mineral crystallization) and/or petrogenetic control (incorporation of uranium from a protolith with fractionated 238U/235U into a parental magma) on 238U/235U fractionation processes that operate at magmatic temperatures.
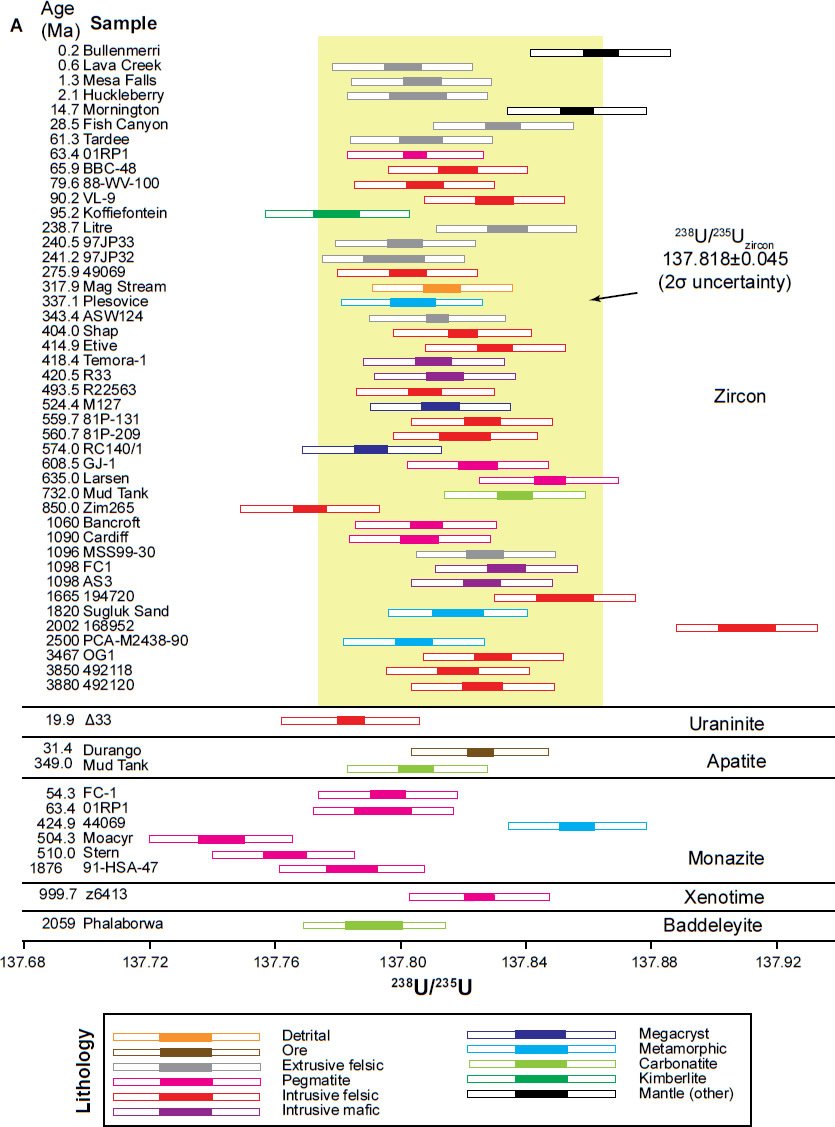
Figure 15. Summary plot of the 238U/235U ratio values of minerals after Hiess et al. (2012), including the 44 samples used to define their recommended 238U/235U ratio value for zircon (represented by the solid yellow band). Solid and open boxes for each sample represent 2σ measured and total uncertainties, respectively.
Of their 45 zircon 238U/235U measurements, Hiess et al. (2012) found that 44 of them were within a range of ~1‰, from 137.772 (zircon from an 850 Ma granite in Zimbabwe) to 137.908 (zircon from a 2002 Ma Gascoyne granite in Western Australia). All five samples of uraninite, apatite, xenotime, and baddeleyite fall within the 238U/235U compositional range of these zircons. Those 44 zircons define an approximately normally distributed population with a mean 238U/235U value of 137.818 and a standard deviation of 0.022. They thus proposed that this average zircon value of 137.818 ± 0.045 (2σ) should be adopted for all zircon U-Pb age determinations, because it reflects the average uranium isotopic composition and variability of terrestrial zircon, and it is broadly representative of the average crustal and “bulk Earth” 238U/235U composition.
Variations in the 238U/235U ratio in meteorites
The 238U/235U ratio has also long been assumed to be invariant at 137.88 in meteoritic and lunar materials (Tatsumoto and Rosholt 1970). Their use has been the cornerstone of Pb-Pb dating, which has supposedly defined the claimed absolute age of the solar system. However, Brennecka et al. (2010b) reported variable 238U/235U ratios for calcium-aluminum-rich inclusions (CAIs) from the Allende carbonaceous chondrite ranging between 137.409 ± 0.039 and 137.885 ± 0.009, which implies substantial uncertainties in their determined Pb-Pb ages. They also found a correlation of the 238U/235U ratios with proxies for curium/uranium (that is, Th/U and Nd/U), which they reasoned provided strong evidence that the observed variations of 238U/235U ratios in the CAIs were produced by the decay of extant curium-247 (247Cm) in the postulated evolutionary scenario for the early solar system. Such seemingly contrived explanations appear to be convenient ways to yet again come up with an unprovable deep-time evolution-based hypothesis to explain obvious discrepancies! Further 238U/235U ratio analyses for a wide variety of meteoritic materials have now been reported by Amelin et al. (2010); Bouvier et al. (2011); Brennecka, Budde, and Kleine (2015); Brennecka and Wadhwa (2012); Connelly et al. (2012); Goldmann et al. (2015); and Spivak-Birndorf et al. (2015).
In their study, Goldmann et al. (2015) combined their 238U/235U ratio analyzes with those from the literature for a total of 39 bulk meteorite samples from different meteorite groups and types (fig. 16). They found the 238U/235U ratios range between 137.711 (Richardton H5) and 137.891 (Elenovka L5), a range of 1.3‰). The largest variations thus appear to occur among the ordinary chondrites. However, the 238U/235U ratios of 20 of the 27 meteorites they analyzed overlap within analytical uncertainties with the narrow range defined by terrestrial basalts (137.778–137.803) (determined by them and by Weyer et al. 2008). Thus they regarded them as the likely best representatives for the 238U/235U composition of the bulk silicate earth. Furthermore, the average 238U/235U ratio of all investigated meteorite groups overlaps with that of terrestrial basalts (137.795 ± 0.013) (fig.16).
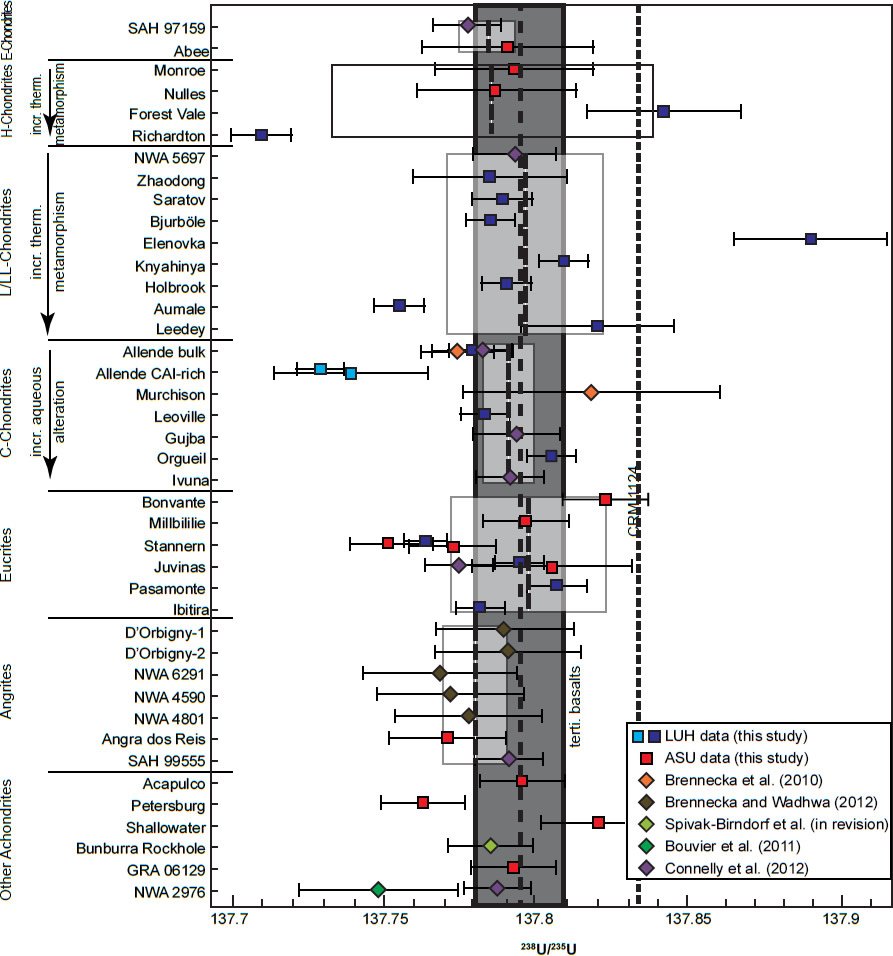
Figure 16. 238U/235U ratios of 39 meteorites, sorted by group (after Goldmann et al. 2015). The 238U/235U ratio of NBL CRM 112a (137.837; Richter et al. 2010) is shown as the vertical dotted line. The average 238U/235U ratio of each meteorite group is shown as a thin vertical dashed line (the surrounding transparent box indicates the calculated one standard deviation uncertainty). The average 238U/235U ratio and the 95% confidence interval of terrestrial basalts relative to the 238U/235U ratio value of NBL CRM 112a (Goldmann et al. 2015; Weyer et al. 2008) is shown as the thick vertical dashed line and dark grey box, respectively. The error bars for individual samples represent the 2σ deviations.
The bulk meteorite samples analyzed by Goldmann et al. (2015) did not show a negative correlation of 238U/235U with Nd/U or Th/U (used as proxies for the Cm/U ratio), as would be expected if radiogenic 235U had been generated by the decay of extant 247Cm in the early solar system. They concluded that the solar system must have a broadly homogeneous 238U/235U composition, because only a limited number of meteorites appear to display detectable variations in 238U/235U ratios. Furthermore, both decay of 247Cm and isotope fractionation are likely responsible for the 238U/235U ratio variations detected in CAIs and ordinary chondrites, respectively.
Therefore, Goldmann et al. (2015) calculated the average 238U/235U ratio of the investigated meteorite groups (including the data compiled from the literature) and the terrestrial basalts as 137.794 ± 0.027 (at a 95% confidence level, including all propagated uncertainties). They thus argued that this value represents the best estimate for the 238U/235U composition of the earth and the solar system. They also argued that this value should now be used for U-Pb and Pb-Pb dating of solar system materials, provided the precise 238U/235U composition of the sample is unknown.
Variations in the 238U/235U ratio in natural materials
Once the experimental protocols for the precise measurement of 238U/235U ratios by MC-ICP-MS were established, some of the earliest investigations of the natural fractionation of 238U/235U were those of Stirling et al. (2007) and Weyer et al. (2008), who analyzed seawater, groundwater, river water, basalts, granites, black shales, corals (both fossil and modern), speleothems, suboxic sediments, manganese nodules, banded iron formations, and uraninites. Tissot and Dauphas (2015) analyzed the 238U/235U ratios in a large number of a wide variety of rock and mineral samples used as 41 geostandards, as well as sea, lake and river waters, corals, oysters, and evaporites. They then combined their data with that available in 32 studies reported in the literature, including the studies of uranium ores and minerals, and of meteorites already discussed. These data are presented in Fig. 17.
Figure 17. Compilation of the δ238U values obtained by Tissot and Dauphas (2015) in their study and from the literature. The top panel (a) shows the modern isotope taxonomy, while the bottom panel (b) shows the data for individual minerals, ore deposits, past sediments, and meteorites and inclusions. Note the different vertical scales of the two panels. The horizontal blue line is the average seawater δ238U value of −0.38 ± 0.010/00 (13 samples). Subsurface modern carbonates include carbonates down to 40 cm below water/sediment interface. Abbreviations: AOC = altered oceanic crust, OAE = oceanic anoxic event, ORM = organic-rich mudrocks, BIF = banded iron formations, OC = ordinary chondrites, CC = carbonaceous chondrites, and EC = enstatite chondrites. Minerals include albite, apatite, baddeleyite, glauconite, biotite, phlogopite, monazite, quartz, titanite, xenotime, zinwaldite, and zircon. Effusive rocks* include andesites, basalts, alkali basalts, rhyolites, and trachytes. Intrusive rocks* include diorites, granites, granodiorites, syenite, and mica-schist. Achondrites include howardites, eucrites, aubrite, and acapulcoites.
Tissot and Dauphas (2015) found that in the modern ocean, strong positive isotopic fractionation (+0.6‰) relative to seawater (average δ238U value of -0.38 ± 0.01‰) is recorded in sediments formed in euxinic/anoxic environments (for example, black shales), while Fe-Mn crusts and evaporites show negative fractionation. Minerals show a much larger range of 238U/235U compositions than crustal rocks, from −0.68‰ to +0.52‰, with a few minerals up to +4.8‰. Using the mass fractions and 238U/235U compositions of various rock types in the earth’s crust, they calculated an average δ238U composition for the continental crust of −0.29 ± 0.03‰, which corresponds to a 238U/235U ratio of 137.797 ± 0.005.
Like so many others who have investigated the variability of the 238U/235U ratio in earth and meteoritic materials, Tissot and Dauphas (2015) discussed the crucial and significant influence of the 238U/235U ratio on Pb-Pb and U-Pb ages, in that they require corrections if the 238U/235U ratio of the sample being dated is unknown. They calculated such corrections for a 1% fractionation in the 238U/235U ratio in a mineral or rock with a 1000 Ma 206Pb model age would require a correction of only 0.015 Ma, whereas the 207Pb and 207Pb/206Pb model ages would require corrections of 0.65 Ma and 2 Ma respectively. Otherwise they recommended their crustal 238U/235U ratio of 137.797 ± 0.005 be used to calculate Pb-Pb and U-Pb ages of continental crust rocks and minerals. This value thus has implications for what value of the 235U half-life should be used in the determination of such Pb-Pb and U-Pb ages, as already discussed.
Is there a trend in the determinations of the 238U and 235U half-lives?
It has already been noted that among the 238U half-life determinations, there is a definite difference between those obtained using ion chambers before 1960, and those obtained in 1960 and 1971 by liquid scintillator and proportional counter respectively (fig. 9). Furthermore, as a result of their critical review and reevaluation, Schön, Winkler, and Kutschera (2004) excluded the Leachman and Schmitt (1957) determination value because “no details” of their experiment were presumably available for assessment of the validity or accuracy of their determination, although Schön, Winkler, and Kutschera (2004) did not provide any further details themselves. They also excluded the Curtiss, Stockman, and Brown (1941) determination value for the same reason (without providing further details), and revised slightly upwards the Schiedt (1935) determination value (apparently to correct for the 235U contribution to the total α-activity). Nevertheless, if the Schiedt (1935) and Leachman and Schmitt (1957) determination values are discounted and excluded as low and high outliers respectively (fig. 9), the rest of the pre-1960 238U half-life values are all definitely higher. Furthermore, the Steyn and Strelow (1960) determination value could be also ignored because of the likelihood their liquid scintillator counted a lower number of α-particles due to scattering in the organic liquid scintillator, and their use of a natural uranium sample which would have introduced difficulties in separating the 238U peak from the overlapping minor 235U peak when analyzing the α-energy spectrum. The latter is a more significant problem, as the range of an alpha in most scintillators is very short. Additionally, the loss of α counts due to the escape of α-particles should have been corrected for if proper counting protocols are used. Thus if the Schiedt (1935), Leachman and Schmitt (1957), and Steyn and Strelow (1960) determination values are ignored, then there is a very definite trend in which the 238U half-life values decreased between 1932 and 1971. The 238U half-life values then leveled out sharply after 1971, primarily because from then until 2016 the results of all critical reviews, reevaluations, and geological age comparisons merely reiterated commitment to the Jaffey et al. (1971) 238U half-life value, even though there were continual calls for more accurate direct counting experimental determinations to be done.
A similar decreasing trend in the determined 235U half-life values is harder to discern (fig. 10). However, if only the direct counting determined 235U half-life values are considered, then there is definitely a slight decreasing trend in the 235U half-life values between 1949 and 1974. Schön, Winkler, and Kutschera (2004) excluded the Kienberger (1949) determination because they considered the experiment and the result “inconsistent” without specifying any reasons or details. Yet even with that Kienberger (1949) 235U half-life value excluded from consideration, the rest of the direct counting determined 235U half-life values between 1950 and 1974 define a subtle decreasing values trend. Furthermore, as with the post-1971 238U half-life values determined by critical review, reevaluation and geological age comparisons, the post-1974 235U half-life values determined by critical review, reevaluation, and geological age comparisons merely reiterated commitment to the Jaffey et al. (1971) 235U half-life value, even though there were continual calls for more accurate direct counting experimental determinations to be done.
It is all too easy to dismiss or downplay the accuracy of the earlier measurements due to the supposedly more rudimentary equipment used in the earlier direct counting experiments. That is what Schön, Winkler, and Kutschera (2004) tended to do in their critical review and reevaluation. For many of the earlier half-life measurements they suggested adjusted half-life values and slightly increased the uncertainties. However, often their adjusted half-life values were higher than the original measurements, so that tends to enhance the decreasing half-life values trend for both 238U and 235U. Furthermore, as stated earlier, the natural tendency for bias towards the most recent measurements as though the more “modern” equipment and methodologies guarantee better results is unfounded, because the earlier experimenters were likely more intimately involved and careful with their equipment and methodologies to obtain excellent results. And since the Jaffey et al. (1971) determined 238U and 235U half-lives are still the recommended values in use today, that at least discounts any overreliance by the early experimenters on computers in their direct counting methodologies. In contrast, the more recent efforts to better determine these half-life values via geological comparisons and critical review reevaluations of all the experimental data have heavily relied on computers, which has resulted in convergence of the recommended half-life values. Thus if we instead treat the earlier experimental results as very reasonable and worthy of acceptance, then these decreasing trends in the 238U and 235U half-life values over the last 70–80 years would appear to be real.
So what would be the significance of these decreasing trends in the 238U and 235U half-life values? Vardiman, Snelling, and Chaffin (2005) reported several lines of compelling evidence for an episode of accelerated nuclear decay in the recent past, when radioisotope decay rates would have been much faster by five orders of magnitude compared to today’s measured rates. This evidence includes many systematically discordant radioisotope dates for the same rock units (Austin 2005; Snelling 2005). However, at the end of that episode the accelerated radioisotope decay rates would not have simply dropped abruptly to their currently measured values. Instead, the radioisotope decay rates would have rapidly decelerated over an extended period of time. Thus an exponential reduction of the decay rates over time would explain their apparent slowdown in recent history. And if the accelerated decay episode was in the recent past, such as during the biblical global Flood cataclysm only 4300 or so years ago as suggested by Vardiman, Snelling, and Chaffin (2005), then we could expect that some of the deceleration of the radioisotope decay rates may have continued even up until the present time. Thus it is entirely possible and reasonable that these subtle decreasing trends we observe in the 238U and 235U half-life values over the last 70–80 years are not only real, but are consistent with the final phase of deceleration of these radioisotope decay rates after the episode of grossly accelerated radioisotope decay rates in the recent past, as suggested by Vardiman, Snelling, and Chaffin (2005).
The implications of these determinations of the 238U and 235U half-lives
Even the conventional geochronology community agrees that the 238U and 235U half-lives have not yet been determined precisely (Schmitz 2012; Schön, Winkler, and Kutschera 2004; Villa et al. 2016). And what bothers the conventional geochronology community is that these imprecisely determined values for the 238U and 235U half-lives result in larger uncertainties in the ages they obtain when they use them in the U-Pb radioisotope methods.
However, this situation is even worse, for several reasons. First, the already well-documented significant widespread variations in the 238U/235U ratio in rocks, minerals, meteorites and other natural materials due to fractionation between the 238U and 235U isotopes not only greatly increases the uncertainties in the 235U half-life value recommended to be used in U-Pb age calculations, but also in all the U-Pb age calculations themselves. These widespread variations could also invalidate the entire methodology by disallowing the use of L’Hôpital’s rule for evaluation of the initial Pb isotope ratios. Indeed, if the 238U/235U ratio is a discontinuous function with time, then L’Hôpital’s rule cannot be used to determine the initial Pb isotope ratios. These uncertainties in all the U-Pb age calculations are also because both the methods for determining the 235U half-life and the equations for calculating all U-Pb ages depend on the 238U/235U ratio being precisely known. The only remedy is to determine the 238U/235U ratio for every rock, mineral or meteorite being U-Pb dated as part of the laboratory routine during the mass spectrometer analyses of them. Second, the tendency of the more recent critical reviews, reevaluations, and geological age comparisons has been to simply reinforce the acceptance of the Jaffey et al. (1971) 238U and 235U half-life values as the recommended values for use in all U-Pb age calculations, and has dissuaded experimenters from attempting new direct counting experiments. And third, because the half-lives of all the other radioisotopes used in radioisotope dating of rocks, minerals, and meteorites have ultimately been determined by cross-calibration against the 238U half-life (Snelling 2014a, b; 2015a, b; 2016), the uncertainties in the 238U half-life are magnified in the determinations of the half-lives of all the other parent radioisotopes and thus the radioisotope ages calculated using them.
Furthermore, by assuming the radioisotope decay rates have always been constant, the conventional geological community has been trying to demonstrate that they are constant, instead of allowing the half-life values determined by the direct counting experiments (which ought to be the preferred method because it actually measures the half-lives directly) to dictate their conclusions. If they did, they would have to admit to four compelling conclusions. First, the 238U and 235U decay rates have likely been decreasing slightly over the last 70–80 years. Second, the 238U and 235U half-lives have not only not been precisely determined, but since the slightly decreasing trend in the 238U and 235U decay rates in the recent past may still be continuing, they may never be able to precisely determine the 238U and 235U half-lives. Third, without precisely known 238U and 235U half-lives, the half-lives of 87Rb, 176Lu, 187Re, 147Sm, and 40K cannot be precisely determined by cross-calibrations. Thus the differences between those half-lives and the 238U and 235U half-lives as also measured in direct counting experiments are likely real, which renders invalid the forced agreements of Rb-Sr, Lu-Hf, Re-Os, Sm-Nd, K-Ar, and Ar-Ar ages with U-Pb ages (Snelling 2014a, b; 2015a, b; 2016). And fourth, all the Rb-Sr, Lu-Hf, Re-Os, Sm-Nd, K-Ar, Ar-Ar, and U-Pb radioisotope ages thus calculated from the imprecisely known 87Rb, 176Lu, 187Re, 147Sm, 40K, 238U, and 235U half-lives simply cannot be trusted to be accurate absolute dates.
Therefore, without accurately known 87Rb, 176Lu, 187Re, 147Sm, 40K, 238U, and 235U decay half-lives, Rb-Sr, Lu-Hf, Re-Os, Sm-Nd, K-Ar, Ar-Ar, and U-Pb radioisotope ages cannot be accurately determined. Thus, all Rb-Sr, Lu-Hf, Re-Os, Sm-Nd, K-Ar, Ar-Ar, and U-Pb dating cannot be used to reject the young-earth creationist timescale, especially as current radioisotope dating methodologies are at best hypotheses based on extrapolating current measurements and observations back into an assumed deep time history for the cosmos, contrary to the actual observable experimentally-determined radioisotope decay data. Furthermore, that same experimental evidence suggests radioisotope decay rates may still have been decelerating in recent decades after the episode of five orders of magnitude acceleration of these radioisotope decay rates during the biblical global Flood cataclysm only about 4300 years ago.
Conclusions
During the last 84 years, numerous determinations of the decay constants and half-lives of 238U and 235U have been made using direct counting experiments and geological age comparisons, as well as by critical reviews and reevaluations of all those determinations. By 1971 the direct counting experiments had provided 238U and 235U half-life values with small questionable uncertainties which in the decades since have been repeatedly recommended as the values to be used in all U-Pb age calculations. All the geological age comparison studies have utilized those recommended values in the U-Pb age calculations used to verify and refine the recommended 238U and 235U half-life values, in spite of the admitted philosophical circularity involved. And the critical reviews and reevaluations have all converged on these same recommended 1971 238U and 235U half-life values because of the meticulous care taken in the 1971 direct counting experiments to comprehensively determine the 238U and 235U half-life values with the lowest possible uncertainties, which then gave those experiments the dominant weight in the calculation of mean values.
Yet at the same time there have been repeated calls for more modern, more accurate direct counting experiments to more precisely determine the 238U and 235U half-lives. This has been because the difficulties of determining precise values for the 238U and 235U half-lives are well documented. In particular, the 238U and 235U peaks in the α-energy spectrum have to be accurately delineated where they overlap and from the background α-particles. Also, secular equilibrium with 234U is assumed, while 234U nevertheless ingrows during the time periods of the experiments, which of necessity have to be long enough to collect statistically large sets of counting data. Furthermore, the 235U half-life is always ultimately determined from the determined 238U half-life by assuming the 238U/235U ratio is constant. And the assumption of a constant 238U/235U ratio is crucial in every U-Pb age calculation. Yet in the last decade significant variations in this crucial 238U/235U ratio have been measured in all natural materials tested, including the rocks, accessory U-bearing minerals, and meteorites that are routinely U-Pb dated.
Of equal great significance alongside all those concerns are the clearly observable trends of decreasing 238U and 235U half-life values obtained from the direct counting experiments between 1932 and 1974, especially those that used the same type of counting instruments, and excluding two divergent outliers. All such experiments should be given the most weight in determining the 238U and 235U half-lives, because in them the numbers of parent 238U and 235U atoms that decay over given time periods are directly counted. Yet to admit that the 238U and 235U decay rates may not have been constant in recent decades is tantamount to admitting that the 238U and 235U half-lives might have been many orders of magnitude shorter several thousand years ago and may never be determined precisely. Furthermore, since the 87Rb, 176Lu, 187Re, 147Sm, and 40K half-lives have all been determined by cross-calibration with the 238U half-life by forced agreement of Rb-Sr, Lu-Hf, Re-Os, Sm-Nd, K-Ar, and Ar-Ar ages respectively with U-Pb ages obtained for the same rocks, minerals and meteorites, none of these decay half-lives are really known accurately.
Therefore, without accurately known 87Rb, 176Lu, 187Re, 147Sm, 40K, 238U, and 235U decay half-lives, Rb-Sr, Lu-Hf, Re-Os, Sm-Nd, K-Ar, Ar-Ar, and U-Pb radioisotope ages cannot be accurately determined or be considered absolute ages. Thus, all Rb-Sr, Lu-Hf, Re-Os, Sm-Nd, K-Ar, Ar-Ar, and U-Pb dating cannot be used to reject the young-earth creationist timescale, especially as current radioisotope dating methodologies are at best hypotheses based on extrapolating current measurements and observations back into an assumed deep time history for the cosmos, contrary to the actual observable experimentally-determined radioisotope decay data. Furthermore, that same experimental evidence suggests that radioisotope decay rates have been decreasing in recent decades. This is consistent with the several lines of impeccable evidence that radioisotope decay rates were accelerated by five orders of magnitude during the year-long biblical global Flood cataclysm, and that after that brief episode the decay rates decelerated. That we may still be detecting the radioisotope decay rates decelerating is likewise consistent with the biblical global Flood cataclysm occurring only about 4300 years ago.
Acknowledgments
The invaluable help of my research assistant in compiling and plotting the data in the color coded age diagrams is acknowledged. The three reviewers are also acknowledged for their helpful comments and input, though the final content of this paper is solely my responsibility. Our production assistant Laurel Hemmings is also especially thanked for her painstaking work in preparing this paper for publication.
References
Abe, M., T. Suzuki, Y. Fujii, M. Hada, and K. Hirao. 2008. “An Ab Initio Molecular Orbital Study of the Nuclear Volume Effects in Uranium Isotope Fractionations.” The Journal of Chemical Physics 129 (16): 164309-1–164309-7.
Abe, M., T. Suzuki, Y. Fujii, M. Hada, and K. Hirao. 2010. “Ligand Effect on Uranium Isotope Fractionations Caused by Nuclear Volume Effects: An Ab Initio Relativistic Molecular Orbital Study.” The Journal of Chemical Physics 133 (4): 044309-01–044309-05.
Amelin, Y., A. Kaltenbach, T. Iizuka, C. H. Stirling, T. Ireland, M. Petaev, and S. B. Jacobsen. 2010. “U-Pb Chronology of the Solar System’s Oldest Solids with Variable 238U/235U.” Earth and Planetary Science Letters 300 (3–4): 343–350.
Apt, K. E., J. P. Balagna, E. A. Bryant, G. A. Cowan, W. R. Daniels, R. J. Vidale, and D. G. Brookins. 1978. “Search for Other Natural Fission Reactors.” Technical Committee Meeting on Natural Fission Reactors, Panel Proceedings Series, IAEA-TC-119/37, 677–689. Vienna, Austria: International Atomic Energy Agency.
Ashton, F. W. 1919. “A Positive Ray Spectrograph.” Philosophical Magazine 38 (series 6): 707–714.
Austin, S. A. 2005. “Do Radioisotope Clocks Need Repair? Testing the Assumptions of Isochron Dating Using K-Ar, Rb-Sr, Sm-Nd, and Pb-Pb Isotopes.” In Radioisotopes and the Age of the Earth: Results of a Young-Earth Creationist Research Initiative. Edited by L. Vardiman, A. A. Snelling, and E. F. Chaffin, 325–392. El Cajon, California: Institute for Creation Research, and Chino Valley, Arizona: Creation Research Society. http://www.icr.org/article/do-radioisotope-clocks-need-repair/.
Banks, P. O., and L. T. Silver. 1966. “Evaluation of the Decay Constant of Uranium 235 from Lead Isotope Ratios.” Journal of Geophysical Research 71 (16): 4037–4046.
Baudin, G., C. Blain, R. Hagemann, M. Kremer. M. Lucas, L. Merlivat, R. Molina, et al. 1972. “Quelques Données Nouvelles sur les Réactions Nucléaires en Chaîne qui se Sont Produites dans le Gisement d’Oklo.” Comptes Rendus de l’Académie des Sciences 275D: 2291–2294.
Begemann, F., K. R. Ludwig, G. W. Lugmair, K. Min, L. E. Nyquist, P. J. Patchett, P. R. Renne, C.-Y. Shih, I. M. Villa, and R. J. Walker. 2001. “Call for an Improved Set of Decay Constants for Geochronological Use.” Geochimica et Cosmochimica Acta 65 (1): 111–121.
Bigeleisen, J. 1996. “Nuclear Size and Shape Effects in Chemical Reactions: Isotope Chemistry of the Heavy Elements.” Journal of the American Chemical Society 118 (15): 3676–3680.
Bodu, R., H. Bouzigues, N. Morin, and J.-P. Pfiffelmann. 1972. “Sur l’Existence d’Anomalies Isotopiques Rencontrés dan l’Uranium du Gabon.” Comptes Rendus de l’Académie des Sciences 275D: 1731–1732.
Boehnke, P., and T. M. Harrison. 2014. “A Meta-Analysis of Geochronologically Relevant Half-Lives: What’s the Best Decay Constant?” International Geology Review 56 (7): 905–914.
Boltwood, B. B. 1907. “On the Ultimate Disintegration Products of the Radioactive Elements.” American Journal of Science 23 (4): 77–88.
Bopp, C. J., C. C. Lundstrom, T. M. Johnson, and J. J. G. Glessner. 2009. “Variations in 238U/235U in Uranium Ore Deposits: Isotopic Signatures of the U Reduction Process?” Geology 37 (7): 611–614.
Bouvier, A., L. J. Spivak-Birndorf, G. A. Brennecka, and M. Wadhwa. 2011. “New Constraints on Early Solar System Chronology from Al-Mg and U-Pb Isotope Systematics in the Unique Basaltic Achondrite Northwest Africa 2976.” Geochimica et Cosmochimica Acta 75 (18): 5310–5323.
Brennecka, G. A., L. E. Borg, I. D. Hutcheon, M. A. Sharp, and A. D. Anbar. 2010a. “Natural Variations in Uranium Isotope Ratios of Uranium Ore Concentrates: Understanding the 238U/235U Fractionation Mechanism.” Earth and Planetary Science Letters 291 (1–4): 228–233.
Brennecka, G. A., S. Weyer, M. Wadhwa, P. E. Janney, J. Zipfel, and A. D. Anbar. 2010b. “238U/235U Variations in Meteorites: Extant 247Cm and Implications for Pb-Pb Dating.” Science 327 (5964): 449–451.
Brennecka, G. A., and M. Wadhwa. 2012. “Uranium Isotope Compositions of the Basaltic Angrite Meteorites and the Chronological Implications for the Early Solar System.” Proceedings of the National Academy of Sciences USA 109 (24): 9299–9303.
Brennecka, G. A., G. Budde, and T. Kleine. 2015. “Uranium Isotopic Composition and Absolute Ages of Allende Chondrules.” Meteoritics and Planetary Science 50 (12): 1995–2002.
Bueno, C. C., and M. D. S. Santos. 1993. “Measurement of the Absolute Activity of 235U by the Generalized Coincidence Method.” Applied Radiation and Isotopes 44 (3): 567–574.
Chaffin, E.F. 2005. “Accelerated Decay: Theoretical Considerations.” In Radioisotopes and the Age of the Earth: Results of a Young-Earth Creationist Research Initiative. Edited by L. Vardiman, A. A. Snelling, and E. F. Chaffin, 525–585. El Cajon, California: Institute for Creation Research, and Chino Valley, Arizona: Creation Research Society. http://www.icr.org/article/accelerated-decay-theoreticalconsiderations/.
Cherniak, D. J., and E. B. Watson. 2003. “Diffusion in Zircon.” In Zircon, Reviews in Mineralogy and Geochemistry. Vol. 53, 113–143. Edited by J. M Hanchar and P. W. O. Hoskin, Washington DC: Mineralogical Society of America.
Clark, F. L., H. J. Spencer-Palmer, and R. N. Woodward. 1957. “The Determination of the Half-Lives and α-Particle Energies of Some Radioactive Isotopes. Part I. The Determination of the Half-Lives of Uranium 235 and Uranium 238.” Journal of the South African Chemical Institute 10: 62–83.
Condon, D. J., N. McLean, S. R. Noble, and S. A. Bowring. 2010. “Isotopic Composition (238U/235U) of Some Commonly Used Uranium Reference Materials.” Geochimica et Cosmochimica Acta 74 (24): 7127–7143.
Connelly, J. N., M. Bizzarro, A. N. Krot, Å. Nordlund, D. Wielandt, and M. A. Ivanova. 2012. “The Absolute Chronology and Thermal Processing of Solids in the Solar Protoplanetary Disk.” Science 338 (6107): 651–655.
Coursol, N., F. Lagoutine, and B. Duchemin. 1990. “Evaluation of Non-Neutron Nuclear Data for the Uranium-238 Decay Chain.” Nuclear Instruments and Methods in Physics Research A 286 (3): 589–594.
Cowan, G. A. 1976. “A Natural Fission Reactor.” Scientific American 235 (1): 36–47.
Cowan, G. A., and H. H. Adler. 1976. “The Variability of the Natural Abundance of 235U.” Geochimica et Cosmochimica Acta 40 (12): 1487–1490.
Curtiss, L. F., L. L. Stockman, and B. W. Brown. 1941. “Intercomparison of Mass Spectrometer and Alpha-Particle Methods for Isotopic Analysis.” U.S. National Bureau of Standards Report No. A-80. Washington, DC: U.S. Government Printing Office.
de Laeter, J. R., J. K. Böhlke, H. Briève, H. Hidaka, H. S. Peiser, K. J. R. Rosman, and P. D. P. Taylor. 2003. “Atomic Weights of the Elements: Review 2000 (IUPAC Technical Report).” Pure and Applied Chemistry 75 (6): 683–800.
Dempster, A. J. 1918. “A New Method of Positive Ray Analysis.” Physical Review 11: 316–325.
Deruytter, A. J., I. G. Schröder, and J. A. Moore. 1965. “Measurement of the Half-Life of U235 for Alpha Emission.” Nuclear Science and Engineering 21 (3): 325–328.
Deruytter, A. J., and G. Wegener-Penning. 1974. “Redetermination of the Half-Life of 235U for α Emission.” Physical Review C 10 (1): 383–385.
Dickin, A. P. 2005. Radiogenic Isotope Geology. 2nd ed. Cambridge, United Kingdom: Cambridge University Press.
Duchemin, B., N. Coursol, and M. M. Bé 1994. “The Re-Evaluation of Decay Data for the U-238 Chain.” Nuclear Instruments and Methods in Physics Research A 339 (1–2): 146–150.
Ewing, R. C., A. Meldrum, L. Wang, W. J. Weber, and L. R. Corrales. 2003. “Radiation Effects in Zircon.” In Zircon, Reviews in Mineralogy and Geochemistry. Vol. 53. Edited by J. M Hanchar and P. W. O. Hoskin, 387–425. Washington DC: Mineralogical Society of America.
Faure, G., and T. M. Mensing. 2005. Isotopes: Principles and Applications. 3rd ed. Hoboken, New Jersey: John Wiley & Sons.
Fleming, E. H., A. Ghiorso, and B. B. Cunningham. 1952. “The Specific Alpha-Activities and Half-Lives of U234 , U235, and U236.” Physical Review 88 (3): 642–652.
Ghiorso, A. 1951. “Complex Alpha-Spectrum of U235.” Physical Review 82 (6): 979.
Goldmann, A., G. Brennecka, J. Noordmann, S. Weyer, and M. Wadhwa. 2015. “The Uranium Isotopic Composition of the Earth and the Solar System.” Geochimica et Cosmochimica Acta 148: 145–158.
Hamer, A. N., and E. J. Robbins. 1960. “A Search for Variations in the Natural Abundance of Uranium-235.” Geochimica et Cosmochimica Acta 19 (2): 143–145.
Hiess, J., D. J. Condon, N. McLean, and S. R. Noble. 2012. “238U/235U Systematics in Terrestrial Uranium-Bearing Minerals.” Science 335 (6076): 1610–1614.
Hoffman, D. L., J. Prytulak, D. A. Richards, T. Elliott, C. D. Coath, P. L. Smart, and D. Scholz. 2007. “Procedures for Accurate U and Th Isotope Measurements by High Precision MC-ICPMS.” International Journal of Mass Spectrometry 264 (2–3): 97–109.
Holmes, A. 1913. The Age of the Earth. London, United Kingdom: Harper and Brothers.
Jaffey, A. H., K. F. Flynn, L. E. Glendenin, W. C. Bentley, and A. M. Essling. 1971. “Precision Measurement of Half-Lives and Specific Activities of 235U and 238U.” Physical Review C 4 (5): 1889–1906.
Keegan, E., S. Richter, I. Kelly, H. Wong, P. Gadd, H. Kuehn, and A. Alonso-Munoz. 2008. “The Provenance of Australian Uranium Ore Concentrates by Elemental and Isotopic Analysis.” Applied Geochemistry 23 (4): 765–777.
Kienberger, C. A. 1949. “The Uranium 234 Content of Natural Uranium and the Specific Alpha-Activities of the Isotopes.” Physical Review 76 (11): 1561–1563.
Knight, B. 1950. “Specific Alpha Activity of U-235.” Oak Ridge National Laboratory Report No. K-663 (unpublished).
Kossert, K., and E. Günther. 2004. “LSC Measurements of the Half-Life of 40K.” Applied Radiation and Isotopes 60 (2–4): 459–464.
Kovarik, A. F., and N. I. Adams, Jr. 1932. “A New Determination of the Disintegration Constant of Uranium by the Method of Counting α-Particles.” Physical Review 40: 718–726.
Kovarik, A. F., and N. I. Adams. 1955. “Redetermination of the Disintegration Constant of U238.” Physical Review 98 (1): 46. Kuroda, P. K. 1982. The Origin of the Chemical Elements and the Oklo Phenomenon. New York: Springer-Verlag.
Lancelot, J. R., A. Vitrac, and C. J. Allègre. 1975. “The Oklo Natural Reactor: Age and Evolution Studies by U-Pb and Rb-Sr Systematics.” Earth and Planetary Science Letters 25 (2): 189–196.
Leachman, R. B., and H. W. Schmitt. 1957. “The Cross-Section for 238U Fission by Fission Neutrons.” Journal of Nuclear Energy 4 (1): 38–43.
Ludwig, K. R. 1993. “Pb Dat 1.24: A Computer Program for Processing Pb-U-Th Isotope Data.” USGS Open-File Report 88-542. Washington, DC: U.S. Geological Survey.
Ludwig, K. R. 1998. “On the Treatment of Concordant Uranium-Lead Ages.” Geochimica et Cosmochimica Acta 62 (4): 665–676.
Ludwig, K. R. 2000. “Decay Constant Errors in U-Pb Concordia-Intercept Ages.” Chemical Geology 166 (3–4): 315–318.
Malyshev, V. I., V. G. Melkov, A. L. Yakubovich, Z. A. Sokolova, L. V. Sumin, M. B. Shiryayeva, M. Ye. Kotsen, et al. 1975. “Tests of the Theory of Possible Shifts of the U235/U238 Ratio in Nature.” Doklady Akademii Nauk SSSR, Earth Sciences Section 223 (2): 212–214.
Mattinson, J. M. 1987. “U-Pb Ages of Zircons: A Basic Examination of Error Propagation.” Chemical Geology 66 (1–2): 151–162.
Mattinson, J. M. 1994a. “Uranium Decay Constant Uncertainties, and Their Implications for High-Resolution U-Pb Geochronology.” Geological Society of America Abstracts with Programs 77: A-221.
Mattinson, J. M. 1994b. “Real and Apparent Concordance and Discordance in the U-Pb Systematics of Zircon: Limitations of ‘High Precision’ U/Pb and Pb/Pb Ages.” EOS, Transactions of the American Geophysical Union 75: 691.
Mattinson, J. M. 2000. “Revising the ‘Gold Standard’, the Uranium Decay Constants of Jaffrey et al., 1971.” EOS, Transactions of the American Geophysical Union 81: abstract S444.
Mattinson, J. M. 2010. “Analysis of the Relative Decay Constants of 235U and 238U by Multi-Step CA-TIMS Measurements of Closed-System Natural Zircon Samples.” Chemical Geology 275 (3–4): 186–198.
Miller, J. L. 2012. “Time to Reset Isotopic Clocks? Two New Studies Revise Key Parameters in Radiometric Dating.” Physics Today 65 (6): 20–22.
Min K., R. Mundil, P. R. Renne, and K. R. Ludwig. 2000. “A Test for Systematic Errors in 40Ar/39Ar Geochronology Through Comparison with U-Pb Analysis of a 1.1 Ga Rhyolite.” Geochimica et Cosmochimica Acta 64 (1): 73–98.
Murphy, M. J., C. H. Stirling, A. Kaltenbach, S. P. Turner, and B. F. Schaefer. 2014. “Fractionation of 238U/235U by Reduction During Low Temperature Uranium Mineralisation Processes.” Earth and Planetary Science Letters 388: 306–317.
Nier, A. O. 1939. “The Isotopic Constitution of Uranium and the Half-Lives of the Uranium Isotopes. I.” Physical Review 55 (2): 150–153.
Nier, A. O. 1940. “A Mass Spectrometer for Routine Isotope Abundance Measurements.” Review of Scientific Instruments 11 (7): 398–411.
Renne, P. R., D. B. Karner, and K. R. Ludwig. 1998. “Absolute Ages Aren’t Exactly.” Science 282 (5395): 1840–1841.
Richter, S., A. Alonso-Munoz, W. De Bolle, R. Wellum, and P. D. P. Taylor. 1999. “Isotopic ‘Fingerprints’ for Natural Uranium Ore Samples.” International Journal of Mass Spectrometry 193 (1): 9–14.
Richter, S., A. Alonso-Munoz, R. Eykens, U. Jacobsson, H. Kuehn, A. Verbruggen, Y. Aregbe, R. Wellum, and E. Keegan. 2008. “The Isotopic Composition of Natural Uranium Samples—Measurements Using the New n(233U)/ n(236U) Double Spike IRMM-3636.” International Journal of Mass Spectrometry 269 (1–2): 145–148.
Richter, S., A. Alonso-Munoz, Y. Aregbe, R. Eykens, F. Kehoe, H. Kühn, N. Kivel, A. Verbruggen, R. Wellum, and P. D. P. Taylor. 2009. “A New Series of Uranium Isotope Reference Materials for Investigating the Linearity of Secondary Electron Multipliers in Isotope Mass Spectrometry.” International Journal of Mass Spectrometry 281 (3): 115–125.
Richter, S., R. Eykens, H. Kühn, Y. Aregbe, A. Verbruggen, and S. Weyer. 2010. “New Average Values for n(238U)/ n(235U) Isotope Ratios of Natural Uranium Standards.” International Journal of Mass Spectrometry 295 (1–2): 94–97.
Riley, G. H., and M. J. Korsch. 1980. “Natural Reactor Studies.” In Uranium in the Pine Creek Geosyncline. Edited by J. Ferguson and A. B Goleby, 407–416. Vienna, Austria: International Atomic Energy Agency.
Rudnick, R. L., and S. Gao. 2005 “Composition of the Continental Crust.” In The Crust. Edited by R. L. Rudnick. Treatise on Geochemistry. Edited by H. D. Holland and K. K. Turekian. Vol. 3, 1–64. Amsterdam, The Netherlands: Elsevier.
Rutherford, E. 1906. Radioactive Transformations. New York: Charles Scribner’s Sons. Sayag, M.G.-J.1951. “Mesure du Rapport des Activites de U235 et U234 dans l’Uranium Naturel a l’Aide d’une Chambre d’Ionisation a Grille.” Comptes Rendus de l’Academie des Sciences Paris 232: 2091–2093.
Schauble. E. A. 2007. “Role of Nuclear Volume in Driving Equilibrium Stable Isotope Fractionation of Mercury, Thallium, and Other Very Heavy Elements.” Geochimica et Cosmochimica Acta 71 (9): 2170–2189.
Schiedt, R. 1935. “Die Zahl der von Uran Emittierten Alpha-Teilchen (The Number of Alpha Particles Emitted from Uranium).” Österr. Akad. d. Wiss., Sitzungsberichte d. mathem.-naturw. Klasse, Abt. IIa, 144 Bd., Heft 5 und 6: 192.
Schmitz, M. D. 2012. “Radiogenic Isotope Geochronology.” In The Geologic Time Scale 2012. Edited by F. M. Gradstein, J. G. Ogg, M. D. Schmitz, and G. M. Ogg. Vol. 1, chapter 6, 115–126. Amsterdam, The Netherlands: Elsevier.
Schoene, B., J. L. Crowley, D. J. Condon, M. D. Schmitz, and S. A. Bowring. 2006. “Reassessing the Uranium Decay Constants for Geochronology Using ID-TIMS U–Pb Data.” Geochimica et Cosmochimica Acta 70 (2): 426–445.
Schön, R., G. Winkler, and W. Kutschera. 2004. “A Critical Review of Experimental Data for the Half-Lives of the Uranium Isotopes 238U and 235U.” Applied Radiation and Isotopes 60 (2–4): 263–273.
Seydoux-Guillaume, A.-M., B. Bingen, J.-L. Paquette, and V. Bosse. 2015. “Nanoscale Evidence for Uranium Mobility in Zircon and the Discordance of U-Pb Chronometers.” Earth and Planetary Science Letters 409: 43-48.
Slodzian, G., and A. Havette. 1974. “Ion Microanalyser Observation of Samples from the Natural Reactor of Oklo: Preliminary Results.” In Advances in Mass Spectrometry. Edited by A. R. West. Vol. 6, 629–636. Barking, Essex, England: Applied Science Publishers.
Snelling, A. A. 2000. “Geochemical Processes in the Mantle and Crust.” In Radioisotopes and the Age of the Earth: A Young-Earth Creationist Research Initiative. Edited by L. Vardiman, A. A. Snelling, and E. F. Chaffin, 123–304. El Cajon, California: Institute for Creation Research, and St. Joseph, Missouri: Creation Research Society. http://www.icr.org/rate/.
Snelling, A. A. 2005. “Isochron Discordances and the Role of Mixing and Inheritance of Radioisotopes in the Mantle and Crust.” In Radioisotopes and the Age of the Earth: Results of a Young-Earth Creationist Research Initiative. Edited by L. Vardiman, A. A. Snelling, and E. F. Chaffin, 393–524. El Cajon, California: Institute for Creation Research, and Chino Valley, Arizona: Creation Research Society. http://www.icr.org/article/isochron-discordances-role-inheritance/.
Snelling, A. A. 2014a. “Determination of the Radioisotope Decay Constants and Half-Lives: Rubidium-87 (87Rb).” Answers Research Journal 7: 311–322. https://answersingenesis.org/geology/radiometric-dating/determination-radioisotope-decay-constants-and-half-lives-rubidium-87-87Rb/.
Snelling, A. A. 2014b. “Determination of the Radioisotope Decay Constants and Half-Lives: Lutetium-176 (176Lu).” Answers Research Journal 7: 483–497. https://answersingenesis.org/geology/radiometric-dating/determination-radioisotope-decay-constants-and-half-lives-lutetium-176/.
Snelling, A. A. 2015a. “Determination of the Radioisotope Decay Constants and Half-Lives: Rhenium-187 (187Re).” Answers Research Journal 8: 93–111. https://answersingenesis.org/geology/radiometric-dating/determination-radioisotope-decay-constants-and-half-lives-rhenium-187/.
Snelling, A. A. 2015b. “Determination of the Radioisotope Decay Constants and Half-Lives: Samarium-147 (247Sm).” Answers Research Journal 8: 305–321. https://answersingenesis.org/geology/radiometric-dating/determination-radioisotope-decay-constants-and-half-lives-samarium-147/.
Snelling, A. A. 2016. “Determination of the Radioisotope Decay Constants and Half-Lives: Potassium-40 (40K).” Answers Research Journal 9: 171–196. https://answersingenesis.org/geology/radiometric-dating/determination-radioisotope-decay-constants-half-lives-potassium-40/.
Spivak-Birndorf, L. J., A. Bouvier, G. K. Bendix, S. Hammond, G. A. Brennecka, K. Howard, N. Rogers, M. Wadhwa, P. A. Bland, P. Spurný, and M. C. Towner. 2015. “Geochemistry and Chronology of the Bunburra Rockhole Ungrouped Achondrite.” Meteoritics and Planetary Science 50 (5): 958–975.
Steiger, R. H., and E. Jäger. 1977. “Subcommission on Geochronology: Convention on the Use of Decay Constants in Geo- and Cosmochronology.” Earth and Planetary Science Letters 36 (3): 359–362.
Steyn, J., and F. W. E. Strelow. 1960. “The Determination of the Half-Life of 238U, Absolute Counting of α Particles in a 4π-Liquid Scintillation Counter.” In Metrology of Radionuclides. Edited by A. Sanielevici, 155–161. Vienna, Austria: International Atomic Energy Agency.
Stirling, C. H., M. B. Anderson, E.-K. Potter, and A. N. Halliday. 2007. “Low-Temperature Isotopic Fractionation of Uranium.” Earth and Planetary Science Letters 264 (1–2): 208–225.
Tatsumoto, M., and J. N. Rosholt. 1970. “Age of the Moon: An Isotopic study of Uranium-Thorium-Lead Systematics of Lunar Samples.” Science 167 (3918): 461–463.
Tera, F., and G. J. Wasserburg. 1972. “U-Th-Pb Systematics in Three Apollo 14 Basalts and the Problem of Initial Pb in Lunar Rocks.” Earth and Planetary Science Letters 14 (3): 281–304.
Tera F., and R. W. Carlson. 1999. “Assessment of the Pb-Pb and U-Pb Chronometry of the Early Solar System.” Geochimica et Cosmochimica Acta 63 (11–12): 1877–1899.
Thomson, J. J. 1911. “A New Method of Chemical Analysis.” Nature 86 (2170): 466–469.
Tissot, F. L. H., and N. Dauphas. 2015. “Uranium Isotopic Compositions of the Crust and Ocean: Age Corrections, U Budget and Global Extent of Modern Anoxia.” Geochimica et Cosmochimica Acta 167: 113–143.
Uvarova, Y. A., T. K. Kyser, M. L. Geagea, and D. Chipley. 2014. “Variations in the Uranium Isotopic Compositions of Uranium Ores from Different Types of Uranium Deposits.” Geochimica et Cosmochimica Acta 146: 1–17.
Vardiman, L., A. A. Snelling, and E. F. Chaffin, eds. 2000. Radioisotopes and the Age of the Earth: A Young-earth Creationist Research Initiative. El Cajon, California: Institute for Creation Research, and St. Joseph, Missouri: Creation Research Society. http://www.icr.org/rate/.
Vardiman, L., A. A. Snelling, and E. F. Chaffin, eds. 2005. Radioisotopes and the Age of the Earth: Results of a Youngearth Creationist Research Initiative. El Cajon, California: Institute for Creation Research, and Chino Valley, Arizona: Creation Research Society. http://www.icr.org/rate2/.
Villa, I. M. 2010. “Disequilibrium Textures versus Equilibrium Modelling: Geochronology at the Crossroads.” In Advances in Interpretation of Geological Processes. Edited by M. I. Spalla, A. M. Marotta, and G. Gosso. Geological Society of London Special Publication 332, 1–15.
Villa, I. M., M. L. Bonardi, P. De Bièvre, N. E. Holden, and P. R. Renne. 2016. “IUPAC-IUGS Status Report on the Half-Lives of 238U, 235U and 234U.” Geochimica et Cosmochimica Acta 172: 387–392.
Wasserburg G. J., D. A. Papanastassiou, E. V. Nemov, and C. A. Bauman. 1969. “A Programmable Magnetic Field Mass Spectrometer with On-Line Data Processing.” Reviews of Scientific Instruments 40 (2): 288–295.
Wetherill, G. W. 1956. “Discordant Uranium-Lead Ages.” Transactions of the American Geophysical Union 37 (3): 320–326.
Wetherill, G. W. 1963. “Discordant Uranium-Lead Ages –Pt. 2. Discordant Ages Resulting from Diffusion of Lead and Uranium.” Journal of Geophysical Research 68 (10): 2957–2965.
Wetherill, G. W. 1966. “Radioactive Decay Constants and Energies.” In Handbook of Physical Constants. Edited by S. E. Clarke. Geological Society of America Memoir 97, 514–519. Boulder, Colorado: Geological Society of America.
Weyer, S., A. D. Anbar, A. Gerdes, G. W. Gordon, T. J. Algeo, and E. A. Boyle. 2008. “Natural Fractionation of 238U/235U.” Geochimica et Cosmochimica Acta 72 (2): 345–359.
White, L. T., and T. R. Ireland. 2012. “High-Uranium Matrix Effect in Zircon and its Implications for SHRIMP U-Pb Age Determinations.” Chemical Geology 306–307: 78–91.
White, P. H., G. J. Wall, and F. R. Pontet. 1965. “A Remeasurement of the Half-Life of 235U.” Journal of Nuclear Energy A/B 19: 33–35.
Winkler, G. 1998. “On the Role of Covariances for Uncertainty Estimates in Radioactivity Measurements.” Applied Radiation and Isotopes 49 (9–11): 1153–1157.
Würger, E., K. P. Meyer, and P. Huber. 1957. “Lebensdauer und Zerfallsschema von U235.” Helvetica Physica Acta 30: 157–162.
Xiaolong, H., and W. Baosong. 2009. “Evaluation of 235U Decay Data.” Applied Radiation and Isotopes 67 (9): 1541–1549.









