
The views expressed in this paper are those of the writer(s) and are not necessarily those of the ARJ Editor or Answers in Genesis.
Abstract
The DNA-protein paradox has long been a point of contention in the origin of life debate. Since nucleic acids (DNA and RNA) are necessary for protein production, and protein carries out nucleic acid production, a primitive cell could not exist without the simultaneous existence of both types of molecules and a system for faithful replication. Faced with this challenging hurdle, evolutionists countered with the RNA World Hypothesis, which proposes that RNA once performed the dual roles of information carrier and catalyst in primitive cells, with those roles eventually being largely handed over to DNA and protein. Admittedly, the RNA World Hypothesis generates its own set of paradoxes that are similarly troubling, some of which are the focus of current origins research. However, aside from the DNA-protein paradox and its fallouts, what is not generally acknowledged is that there are a plethora of other distinct paradoxes in cell and molecular biology that present equally insurmountable obstacles to the naturalistic origin of life. The purpose of this review is to reveal these paradoxes, describe their significance, and evaluate the rebuttals that exist from the scientific community.
Introduction
As the field of molecular biology bloomed in the mid-20th century and its basic principles became apparent, a clear problem presented itself. DNA is the key information carrier in cells, from which the instructions to make proteins are derived; similarly, the tasks needed to replicate, repair, and process DNA are performed almost exclusively by proteins. DNA and proteins are so interdependent on each other for their existence that it is inconceivable to think that one type of molecule could have existed even temporarily without the other. This problem is known as the DNA-protein paradox (fig. 1), and finding a solution to it was of considerable importance to evolutionists in crafting a reasonable origin-of-life model. In the late 1960s, prominent molecular biologists agreed that nucleic acids were more likely than proteins to be capable of self-replication and began to propose a model in which RNA played a prominent role in early cells. Francis Crick was especially intrigued by this RNA-centric model, remarking that in modern cells, “tRNA looks like Nature’s attempt to make RNA do the job of a protein,” then later, “It is tempting to wonder if the primitive ribosome could have been made entirely of RNA” (Crick 1968) (emphasis in original). In a series of papers, Crick, as well as Carl Woese and Leslie Orgel, fashioned the idea that RNA predated both DNA and protein in primitive cells, with various forms functioning as information carriers and likely also possessing catalytic functions (Crick 1968; Orgel 1968; Woese 1967). Interest in this idea increased dramatically when in the early 1980s, specific RNAs (termed ribozymes) were found to have limited catalytic functions in living cells, possessing either self-splicing activity (Kruger et al. 1982) or the ability to cleave tRNA precursors (Guerrier-Takada et al. 1983). The term “RNA world” was coined soon thereafter (Gilbert 1986).
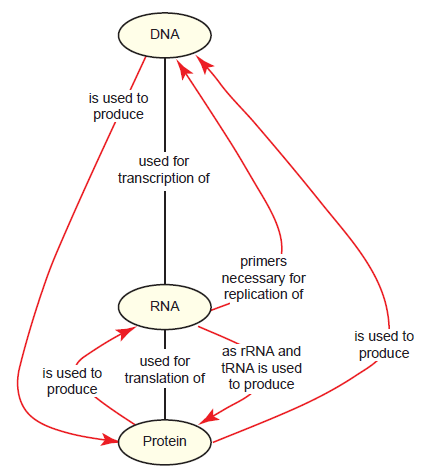
Fig. 1. The DNA-protein paradox as it relates to the central dogma of molecular biology. Relationships between concepts (yellow ovals) are shown by linking them together with lines containing propositional statements. Black lines are primarily for context and should be read from top to bottom; red lines with arrowheads emphasize a statement that is an important component of the paradox. This system is used throughout the paper.
Today, most origins researchers remain committed to the RNA world hypothesis, but it is safe to say that the honeymoon period has long been over. The idea is riddled with conceptual hurdles and defiance of known chemical laws, and though creationists have taken part in the debate, these hurdles have been largely pointed out by materialists themselves. The most significant issues fall into two general categories: the prebiotic synthesis of RNA and the self-replication of RNA. As Leslie Orgel pointed out, “It may be claimed, without too much exaggeration, that the problem of the origin of life is the problem of the origin of the RNA World” (Orgel 2004). Although the RNA world hypothesis serves as an attempted rebuttal to the classic DNA-protein paradox, considering how an RNA world may have originated is paradoxical in and of itself, as is described next.
Fallouts from the DNA-Protein Paradox: Other Paradoxes Emerge
The Homochirality Paradox and the Prebiotic Synthesis of RNA
The RNA world hypothesis requires the non-enzymatic assembly of short RNAs and their stability in a harsh prebiotic environment. Even if this were plausible, a larger problem looms: Where did all of the monomers come from? Even the most talented modern chemists have had little success (at best) at generating nucleotides or their individual components (e.g. purines) non-enzymatically (Orgel 2004). On top of this, the pool would need to consist of pure β-D-nucleotides instead of a racemic mixture, since nucleic acids are one of several types of biomolecules that exhibit homochirality in living systems (Blackmond 2010; McCombs 2014; Sarfati 2000; Swee-eng 1996). Interestingly, this creates a new paradox: Homochirality requires preexisting homochirality (fig. 2). A recent review (McCombs 2014) frames this problem nicely:
Here is the dilemma for evolutionists: because chirality is a physical property, the factors responsible for a chemical reaction to occur are different than the factors responsible for generating new chirality. In the laboratory, the formation of new chirality in a molecule would require a chemical controller to direct and control the chemical reaction, select reactants that contain preexisting homochirality, and perform the reaction so that the creation of a plane of symmetry is avoided. In a natural setting (the evolutionary hypothesis), there is no chemist, and there is no preexisting homochirality, meaning that life with homochirality cannot ever form by natural processes.
Naturally, chemists have developed sophisticated models to try to explain how the biased production of one enantiomer (of a nucleic acid or amino acid) over another could have initiated in a racemic, prebiotic environment; and importantly, how this biased production could have progressed to eventually create the biochemical reality of homochirality that exists today. A popular model suggests that in the prebiotic world, not only could each of the enantiomers undergo autocatalysis, but the reactions producing enantiomers of opposite types could mutually antagonize each other, greatly amplifying one enantiomer over another assuming even minor imbalances existed in the prebiotic pool (Blackmond 2010). But how could these imbalances have originated in the first place? Strangely, some point to meteors as a potential source of enantiomeric excesses in the prebiotic world (McCombs 2014), essentially punting the issue of the origin of homochirality to an obscure astrochemical system that must exist somewhere out there.
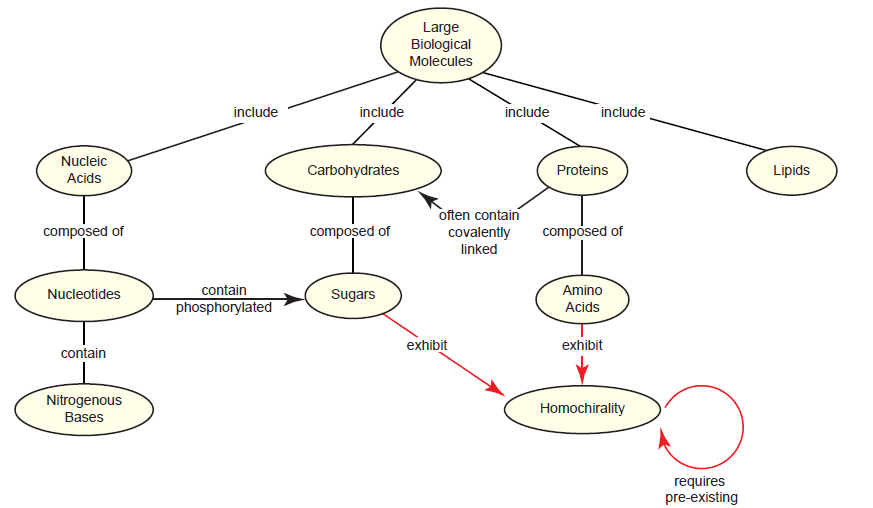
Fig. 2. The homochirality paradox as it relates to large biological molecules. Three out of the four classes of large biological molecules—nucleic acids, carbohydrates, and proteins—exhibit homochirality in living systems because they contain sugars, amino acids, or both. As indicated by the looping arrow, synthesizing homochiral molecules (in living systems or artificially by chemists) requires pre-existing homochirality.
The Self-Replication of RNA: The Ribozyme Paradox and the Folding Paradox
The RNA world hypothesis hinges on the assumption that with the right nucleotides present, RNA could spontaneously assemble and, eventually, a self-replicating ribozyme would emerge. A recent review on the supposed origin of the RNA world (Robertson and Joyce 2012) makes a candid admission:
The previous discussion has tried mightily to present the most optimistic view possible for the emergence of an RNA replicase ribozyme from a soup of random-sequence polynucleotides. It must be admitted, however, that this model does not appear to be very plausible. The discussion has focused on a straw man: The myth of a small RNA molecule that arises de novo and can replicate efficiently and with high fidelity under plausible prebiotic conditions. Not only is such a notion unrealistic in light of current understanding of prebiotic chemistry . . . , but it should strain the credulity of even an optimist’s view of RNA’s catalytic potential.
Later, while pondering how a self-replicating ribozyme (a lengthy RNA with considerable sequence restraints) could emerge from random-sequenced RNAs, Robertson and Joyce unearth a new paradox:
But here one encounters another chicken-and-egg paradox: Without evolution it appears unlikely that a self-replicating ribozyme could arise, but without some form of self-replication there is no way to conduct an evolutionary search for the first, primitive self-replicating ribozyme.
We’ll call this the ribozyme paradox (fig. 3), as it highlights the centrality of the emergence of self-replicating ribozymes to the RNA world hypothesis. But there is another confounding issue as well: A polymerase ribozyme (assuming it exists) can’t just copy itself, but requires a template; thus, in order for the “first” polymerase ribozyme to copy itself, another copy would have to already exist that could be used as a template (Higgs and Lehman 2015). Other theoretical models for RNA replication exist (Joyce 2002), but template-directed nucleic acid polymerization is a hallmark of all known living systems, so to explain away this issue is no small feat.
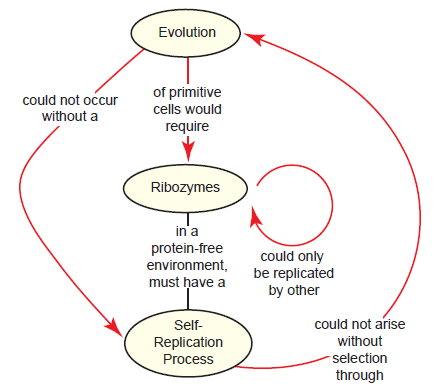
Fig. 3. The ribozyme paradox as it relates to the supposed materialistic origin of life. In the RNA world view of origins, the first cell would have to contain polymerase ribozymes capable of undergoing self-replication. Since replication requires a template, the ribozyme could not even copy itself unless there was another copy already in existence (one to act as the polymerase ribozyme and the other to act as a template for replication), begging the question of how the first replication event could have occurred. This self-replication process could only arise through a search among a pool of RNA molecules for ones that promote replication, but without a self-replication process already intact, it would be impossible for evolution to pursue the first ribozyme.
Given the a priori commitment of many to the RNA world hypothesis, solving this paradox has been a natural focus of origins research. Two potential answers—both highly theoretical—have been suggested as to how RNA evolution could have been kick-started without ribozymes. The first involves non-enzymatic template-directed synthesis, and the second abandons the notion that the RNA world even brought about ribozymes, essentially that “RNA replication arose in the context of an evolving system based on something other than RNA” (Robertson and Joyce 2012). In this view, the RNA World was really just an intermediate operational form of life, preceded by yet another unknown type of system that successfully jumped the hurdles of self-assembly and self-replication. In other words, the RNA world was really just “RNA Later” (Fry 2011; Orgel 2004; Robertson and Joyce 2012).
Further considering the problems with ribozyme self-replication brings about yet another paradox, as presented in the abstract of a recent paper (Ivica et al. 2013):
The hypothesized dual roles of RNA as both information carrier and biocatalyst during the earliest stages of life require a combination of features: good templating ability (for replication) and stable folding (for ribozymes). However, this poses the following paradox: well-folded sequences are poor templates for copying, but poorly folded sequences are unlikely to be good ribozymes.
We’ll call this the folding paradox (fig. 4). It is based on the reasonable assumption that in order for a ribozyme to be functional, it must be stably folded into a complex structure, much like a “modern day” enzyme. However, in the absence of a set-aside genome, this same ribozyme would also need to serve as a template for its own replication. Templates cannot have complex, stable folding if they are to be accessible to free nucleotides and an RNA primer. Clearly, the same RNA cannot meet these two mutually exclusive requirements simultaneously.
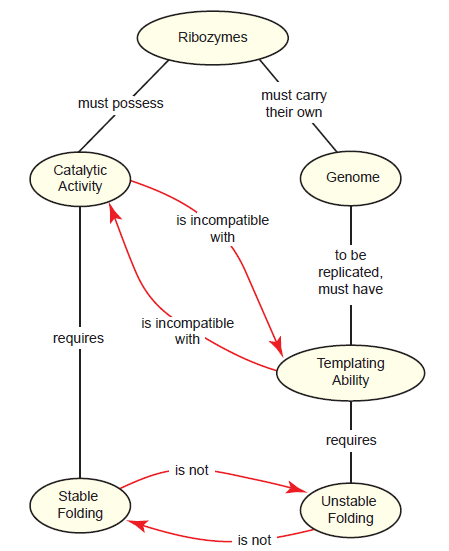
Fig. 4. The folding paradox as it relates to the dual roles of ribozymes in the RNA world. In the RNA world hypothesis, ribozymes necessarily must have dual roles—they must have catalytic activity while simultaneously being able to act as a template for replication. Catalytic activity would require stable folding into complex structures, whereas its role as a template requires unstable, relaxed folding. These two folding states are mutually exclusive, creating a paradox.
Once again, origins researchers have turned to theoretical models and simulations to reason out of this paradox. Using non-enzymatic template-directed synthesis as the assumed replication method, they suggested that a double-stranded ribozyme could use one RNA strand as the actual ribozyme, and the other strand could function as the genome. The ribozyme strand would be well folded, but if it utilized G:U wobble pairing instead of only obeying Watson-Crick pairing rules, the genome strand (a reverse complement sequence) would be less stable due to C:A mispairing. As a result, the genome strand would be in an acceptable conformation to serve as a replication template (Ivica et al. 2013). Though this may seem reasonable on the surface, this model admittedly ignores an equally daunting problem: In the absence of other specialized ribozymes or excessive heat, how could the two strands of the ribozyme faithfully separate in the first place?
Leaving the RNA World
If nothing else, the previous sections should highlight the RNA world hypothesis as a largely theoretical concept that falls short of solving the DNA-protein paradox, even within a materialistic framework. Addressing all of the problems is beyond the scope of this review, and detailed reviews exist elsewhere (Higgs and Lehman 2015; Joyce 2002; Müller 2006; Orgel 2004; Robertson and Joyce 2012; Swee-eng 1996). The point has been to establish the DNA-protein paradox as a model to which other paradoxes in cell and molecular biology can be compared. The reality is that a plethora of unrelated, yet equally weighty paradoxes exist that compounds the problems with the naturalistic view of the origin of life. It is these “other” paradoxes that we will focus on for the remainder of this review.
A “Clutch” of Other Chicken-Egg Paradoxes
The paradoxes discussed below have been selected based on their complexity and centrality to the molecular biology of the cell. They are presented in a logical, but not absolute, order. We’ll start by considering histones, which play a fundamental role in eukaryotic gene expression.
Two Histone Paradoxes
Once considered “boring” proteins that merely provide structural support for DNA (Conova 2003; Marino 2007), histones are now well understood to be central to the dynamic activities of chromatin structure and gene expression in eukaryotic cells. Each core histone is comprised of eight subunits (two each of H2A, H2B, H3, and H4) of which DNA wraps around ~1.7 turns (fig. 5). This DNA-histone complex (147 bp) plus a linker sequence (~53 bp) comprise the most basic unit of chromosome structure, the nucleosome. Each histone subunit contains an N-terminal tail, which protrudes from the core and interacts with a host of proteins, such as nearby histones, or proteins involved in chromatin modification or gene expression. These interactions are regulated through a complicated and vaguely understood series of covalent modifications to the histone tails, which collectively function as the histone code.
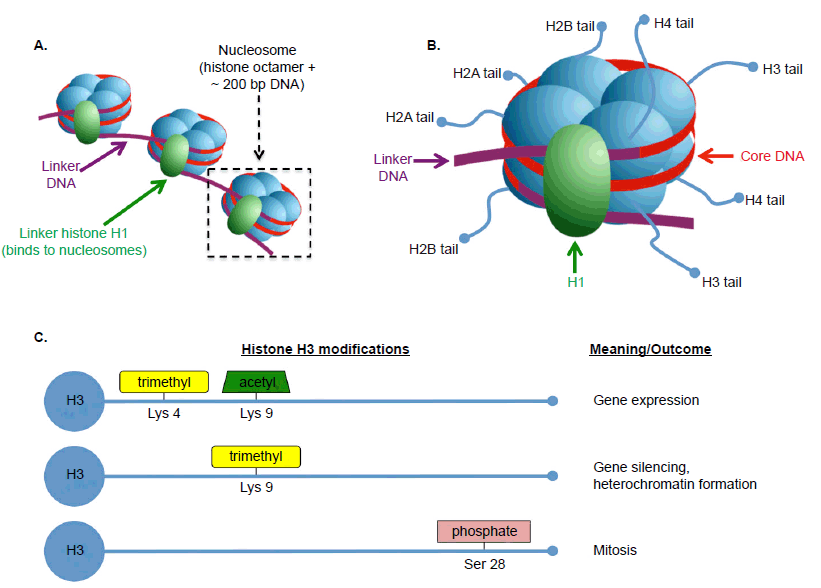
Fig. 5. Basics of histone structure and function. A. Eukaryotic DNA typically exists in the form of chromatin, the basic unit of which is the nucleosome. A nucleosome consists of an octamer of core histones plus ~200 bp of DNA including linker DNA (~53 bp). Histone H1 is not part of the nucleosome itself, but binds to linker DNA and helps stabilize the 30-nm chromatin fiber. B. The core histones consist of two copies each of H2A, H2B, H3, and H4, each of which has a tail domain available for interactions with other proteins. C. Examples of the histone code for histone H3. Certain types of amino acid modifications at certain locations are linked to particular outcomes in the cell. A-B are adapted from Wikimedia Commons (https://commons.wikimedia.org/wiki/File:Nucleosome_organization.png, by Darekk2) under Creative Commons license. Data for C referenced from Alberts et al, Molecular Biology of the Cell 6th edition and http://www.activemotif.com/documents/1815.pdf. Abbreviations: Lys, lysine; Ser, serine.
A consideration of histones brings a paradox that is problematic for evolution: Histones are critical for virtually all processes involving the organization and utilization of DNA, yet these histones are encoded by the very DNA they regulate. We’ll call this the DNA-histone paradox (fig. 6A). Given the centrality of histones to molecular genetics, this has been a topic of interest in the literature. It is proposed that they first appeared in a common ancestor to both Archaea and eukaryotes (Koster, Snel, and Timmers 2015) as a protein to protect DNA from thermal denaturation (Malik and Henikoff 2003). Since the evolution of eukaryotes required a substantial genome enlargement, histones further evolved to facilitate the compaction of the genome (Turner 2014) and to allow the enlarged genome to fit into a small nucleus (Pusarla and Bhargava 2005). The four families of core histones seen in modern eukaryotes were derived from three gene duplication events before the eukaryotic lineage diversified (Mariño-Ramírez, Jordan, and Landsman 2006; Pusarla and Bhargava 2005). Later, but still early in the eukaryotic lineage, histones further acquired the ability to regulate gene expression as the complexity of this process increased (Koster, Snel, and Timmers 2015). The evolution of histone variants (not discussed in detail here) would have been a key step in enabling histones to participate in higher-order regulation of chromatin structure and gene expression, and using them to demarcate chromosomal regions such as centromeres (Pusarla and Bhargava 2005).
This sequence of events may sound reasonable on the surface, but it falls into the same trap as many evolutionary narratives. It is derived almost entirely from comparative sequence data of histones in modern day organisms. By analyzing sequence data (observational science) and reading evolution into it (historical science) with the presupposition of evolution, it is impossible to arrive at any other conclusion. So the story of histone evolution is only explanatory within an evolutionary framework.
A second paradox concerning histones is this: Histone tail modifications regulate gene expression; yet the states of these tail modifications are the result of yet other gene expression events in the cell. We’ll call this the histone tail-gene expression paradox (fig. 6B). The literature does not address this issue directly, except to say that histone tails were not an early feature of histones; they were added later to facilitate genome compaction (Malik and Henikoff 2003). The evolution of the histone code itself is not addressed at any depth. This is not surprising, considering that the histone code is so complex and is read and modified by a plethora of other proteins such as transcription regulatory proteins and chromatin remodeling complexes, which would have to co-evolve with histone tails in order for them to be of much use to the cell. Complexity is the enemy when it comes to concocting an evolutionary storyline.
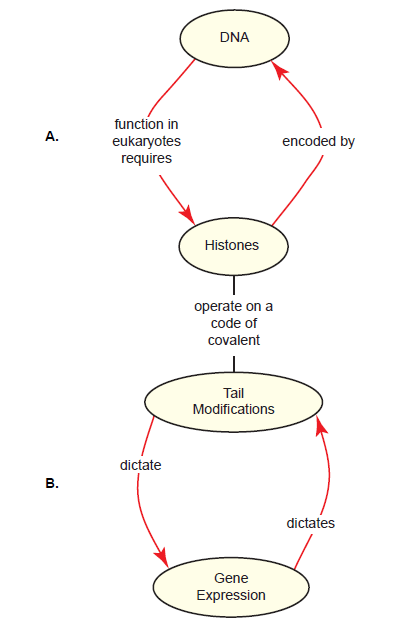
Fig. 6. The histone-DNA (A) and histone tail-gene expression (B) paradoxes. A. In eukaryotes, histones play critical roles in DNA-centric processes DNA such as chromatin dynamics, chromosome separation during mitosis, and gene expression. The information to make these histones is encoded by the DNA itself, creating a paradox, since DNA is unusable in eukaryotes without histones already in place. B. Most functions of histones are an outcome of a histone code, a series of specific modifications of core histone tails (see fig. 5D as an example). The initiation of gene expression relies on specific tail modifications; however, the state of tail modifications is an outcome of yet prior gene expression decisions that were themselves an outcome of the histone code, etc. The interdependency of histone tail modifications and gene expression is paradoxical when one considers how an evolutionarily intermediate system could have existed.
The Eukaryotic Gene Expression Paradox
Nowhere is the complexity of the cell better exhibited than in the expression of the typical eukaryotic gene. Described as “bewilderingly complex” by the leading textbook in molecular biology (Alberts et al. 2015, 384), the decision as to which genes to express and to what extent—and in what contexts—underlies virtually all cellular processes. Ignoring the many intricate regulatory mechanisms for now, gene expression culminates in the recruitment of RNA polymerase II to a gene’s promoter, where it utilizes the information in the DNA to create an RNA transcript, which later may be used by a ribosome to construct a protein. Although RNA polymerase II is complex in and of itself (consisting of 12 subunits in mammals), it is actually part of a much larger protein machine that assembles at the promoter and functions to decide whether and to what extent a gene will be expressed (fig. 7). The decision is ultimately made by context-specific transcription regulatory proteins, which collectively function as a committee and cast up or down votes for gene expression, often by competing for binding sites at cis-regulatory sequences in the vicinity of the gene. These inputs by transcription regulatory proteins cannot be perceived directly by RNA polymerase II; instead, a massive Mediator complex (comprised of over 30 subunits in mammals) coordinates these inputs and regulates the formation of a pre-initiation complex, consisting of six general transcription factors (27 subunits total) and, ultimately, RNA polymerase II (Bourbon 2008; Conaway and Conaway 2011; Yin and Wang 2014). Only upon formation of this complex can transcription occur.
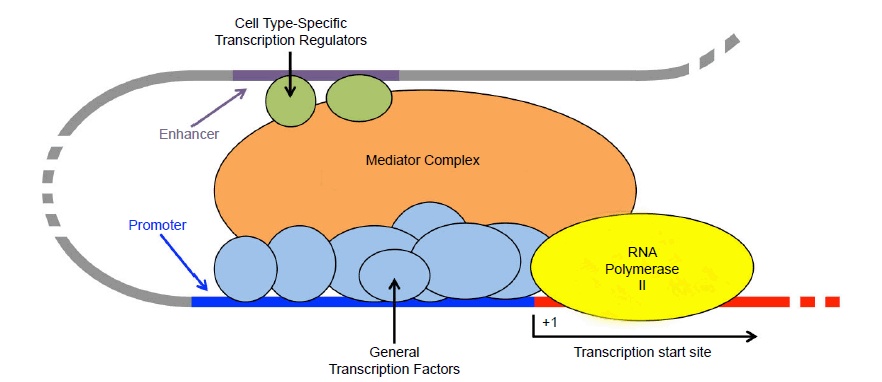
Fig. 7. Eukaryotic transcription initiation via RNA polymerase II. Although RNA polymerase II ultimately transcribes protein-coding genes and many small RNAs, a committee of proteins is required to initiate transcription. General transcription factors bind to the promoter, located directly upstream of the transcription start site (+1) and interact with RNA polymerase II. Cell type-specific transcription regulatory proteins, many of which bind to enhancer elements, are typically key in deciding whether or not a gene is expressed in specific cell types. Enhancers are frequently located many thousands of bases upstream of the gene (specified the dotted gray line). Through DNA looping, these upstream regulators interact with the mediator complex, which coordinates the assembly of the entire transcription initiation complex at the promoter.
The complex process just described is universal to protein-coding genes across eukaryotes. Since a different gene encodes each subunit of the transcription machinery, the reality is that at over 100 gene products (i.e. polypeptide subunits) must coordinate together near a promoter to facilitate the expression of a single gene (Alberts et al. 2015, 313). This includes each of the genes encoding subunits of the transcription machinery itself, bringing about a paradox: The expression of all eukaryotic protein-coding genes requires RNA polymerase II, Mediator, and the general transcription factors, yet each is required for the expression of itself and each of the others (fig. 8). Since gene expression is considerably less complex in prokaryotes and no intermediate system between prokaryotes and eukaryotes exists today, this poses a problem for the evolution of eukaryotic gene expression.
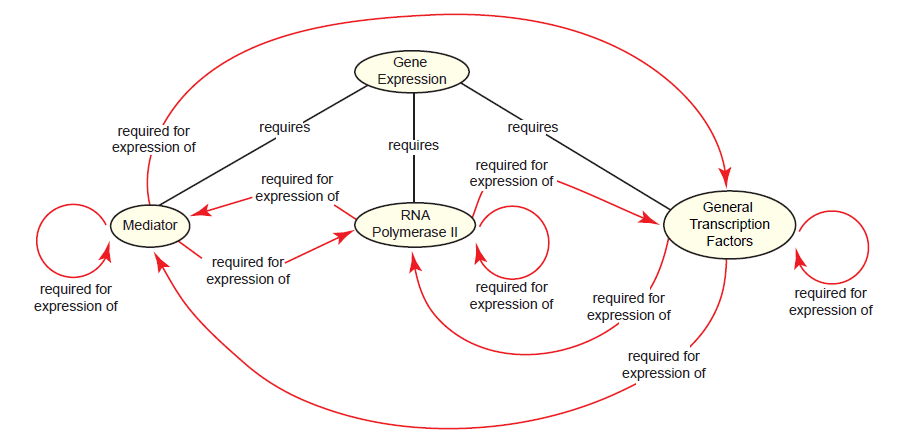
Fig. 8. The eukaryotic gene expression paradox. The expression of protein-coding genes in eukaryotes requires cooperation between the Mediator complex, RNA polymerase II, and the general transcription factors. Transcription regulators and chromatin-modifying proteins are also required, but are not shown here for the sake of clarity. Since all of these components are proteins, each is required for its own expression and for each of the others’ expression.
As expected, most of the studies addressing this issue have utilized comparative genomics within the framework of evolution. Strikingly, it has been proposed that even the most ancient of eukaryotes had a mediator complex of 17 subunits, which rapidly expanded to at least 26 subunits during early eukaryotic evolution. Evolution of mediator would have been largely in place as eukaryotic diversification increased, evidenced by the fact that all 33 known subunits of mediator in humans have counterparts in slime molds, with plants only lacking two of these counterparts (Bourbon 2008). In other words, there is no evidence that a eukaryote did or could have existed with anything but a greatly complex gene expression system already in place. Prokaryotic transcription regulators are believed to have evolved from nucleoid-associated proteins (Visweswariah and Busby 2015) and diversified largely via gene duplication events (Babu and Teichmann 2003); but like the mediator complex, many classes of transcription regulators were fixed early in eukaryotic evolution, with a greater repertoire being added to allow for more complex developmental programs (de Mendoza et al. 2013; Cheatle Jarvela and Hinman 2015). It has even been suggested that most of the “embellishments” present in eukaryotic gene expression evolved in an “arms race” to protect the cell against parasitic nucleic acids (Madhani 2013). However, no mainstream, stepwise transition between the prokaryotic and eukaryotic gene expression systems has been proposed.
A Paradoxical Relationship Between Mutation and DNA Repair
The importance of organizing and utilizing the information in DNA is clearly demonstrated by the effort exerted by the cell in chromatin organization and regulation of gene expression. However, this is all for naught if the integrity of the information encoded by DNA is not preserved. The environment can bombard the cell with DNA-damaging agents such as radiation and chemicals, the extent of which can vary dramatically. However, a more sobering reality is that the normal chemical reactions in a cell can pose a threat to DNA (Lindahl and Barnes 2000). In a typical mammalian cell, this amounts to over 30,000 lesions per day that guarantee a rapid loss of integrity if left unchecked (Alberts et al. 2015, 267). Clearly, DNA repair mechanisms are vital; and since the means by which DNA is damaged are numerous, so must a cell possess diverse processes to repair diverse types of damage.
Though all organisms have DNA repair mechanisms, how and to what extent DNA damage is repaired is uniquely suited to each organism. For example, certain photosynthetic cyanobacteria possess multiple genes dedicated to correcting pyrimidine photodimers resulting from UV light, whereas these genes are lacking in organisms with minimal light exposure such as pathogenic bacteria and hyperthermophilic archaea (Aravind, Walker, and Koonin 1999). The extent of DNA repair also appears to be tied to the benefit a species may potentially gain from mutation (fig. 9). Parasitic bacteria have comparatively minimal repair mechanisms since a population may need to quickly adapt to elude the host’s immune system (Aravind, Walker, and Koonin 1999). Free-living bacteria tend to have more exquisite repair mechanisms, but are still tolerant of considerable mutation. Eukaryotic cells, on the other hand, have the most extensive regulatory mechanisms for DNA repair, often coupling repair to transcription (as do many bacteria) (Hanawalt and Spivak 2008), the cell cycle, and chromatin dynamics. These and many other processes are connected to DNA repair through a centralized DNA damage response (Ciccia and Elledge 2010). In humans, the end result is that despite the persistent internal and external threats to DNA integrity, less than 0.02% of these random mutations escape repair and become permanent (Alberts et al. 2015, 266).
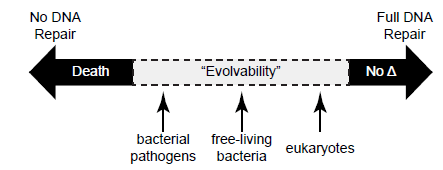
Fig. 9. The spectrum of DNA repair across diverse organisms and its effects on genetic change. DNA repair is necessary to preserve the integrity of DNA and to sustain life, and little to no repair (or alternatively, excessive damage) results in death. All organisms possess DNA repair systems, but none repair with 100% accuracy. Therefore, genetic change via mutations (the so-called raw ingredients for natural selection) occurs over time, providing to organisms what is often referred to as “evolvability.” One creationist perspective is that this quality of “evolvability” provides a mechanism for adaptation.
Clearly, DNA repair is a vital part of the everyday life of the cell. But this brings about a paradox: Evolution requires a steady supply of mutations, but without an already intact DNA repair system, the rapid rate of mutation would be lethal (fig. 10). A system for DNA repair, which is therefore required for evolution, could itself not evolve without a DNA repair mechanism already in place. This is a paradox that really gets at the heart of evolutionary beliefs. Evolutionists accept all of the basic premises, that (1) mutation is the raw material for evolution, (2) excessive mutation rates are lethal, (3) DNA repair holds mutation in check, (4) DNA repair is necessary for life. However, they of course disagree with the main conclusion, that DNA repair could not evolve in an entity without pre-existing repair mechanisms already in place. Starting with the conviction that evolution is true, their question is not whether DNA repair evolved but instead how it evolved. But in doing so, they are forced to make an erroneous exception to the fourth premise (DNA repair is necessary for life), and believe that early life forms must have fared well without effective repair systems. This flies in the face of what we know about all living things, and is neither testable nor falsifiable.
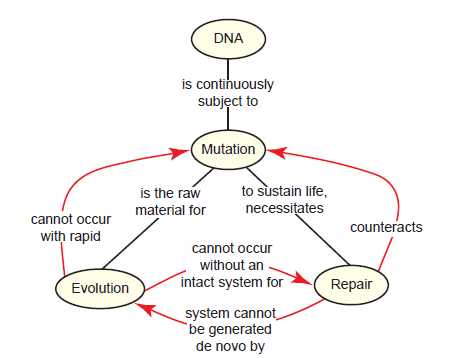
Fig. 10. The mutation-repair paradox. Since internal and external factors cause DNA mutation, an effective repair system is necessary for life. For Neo-Darwinian evolution to occur, mutations are necessary but at a low enough rate to not kill the organism. Therefore, DNA repair is necessary for evolution. Even a basic system for DNA repair could not evolve without an already existing DNA repair system, since mutations would accumulate too rapidly.
The conventional storyline for the evolution of DNA repair brings us back to the RNA world. It is speculated that an RNA-based ancestral cell, also possessing a small set of RNA-interacting proteins, recruited some of these proteins for DNA repair as DNA emerged onto the scene. Further protein evolution resulted in a basic set of ~10 protein domains involved in repair being present in the DNA-based cenancestor (last universal common ancestor or LUCA) (Aravind, Walker, and Koonin 1999). Through a host of evolutionary events including gene duplication/diversification, domain shuffling, horizontal gene transfer, and lineage-specific gene loss, diverse repair mechanisms were able to evolve across all three kingdoms (Aravind, Walker, and Koonin 1999; Koonin 2006; O’Brien 2006) with intricate connections to DNA replication (Koonin 2006). Archaea have the most basic repair systems, with many species supposedly acquiring key repair genes from bacteria through horizontal gene transfer (Koonin 2006). Eukaryotes also acquired many repair genes from bacteria since—once endosymbiosis had supposedly occurred—the bulk of genes from the former bacterial (now mitochondrial) genome were transferred to the nucleus (Aravind, Walker, and Koonin 1999; Koonin 2006). With the emergence of eukaryotic cells came more elaborate cellular processes in which it would be advantageous to link with DNA repair, necessitating the recruitment of many more proteins involved in repair. The rise of multicellularity further amplified this need (Aravind, Walker, and Koonin 1999; Koonin 2006). As always, this evolutionary storyline is derived largely from phylogenomic analyses. It is a testament to man’s creativity, but is fruitless outside of the framework of evolution since it uses data from modern day organisms to speculate about hypothetical, ancient organisms (which never existed).
The Chaperones Paradox
Most newly synthesized polypeptides begin to fold even as they emerge from the ribosome. This is possible because the higher order structures of a polypeptide are largely dictated by its sequence of amino acids. Folding is inevitable as a search ensues for the most energetically favorable conformation. This process typically involves concealing hydrophobic resides into the interior of the polypeptide, binding to cofactors, undergoing covalent modifications such as glycosylation, and ultimately binding to other polypeptides as part of a protein complex (Alberts et al. 2015, 353–354). It is an intricate and slow process for most proteins, and the stakes are high. Newly synthesized polypeptides never exist in isolation, and since they can persist in intermediately folded states with exposed hydrophobic residues for considerable time, they will aggregate if left to themselves (Dobson 2004; Hartl, Bracher, and Hayer-Hartl 2011; Hartl and Hayer-Hartl 2009). Therefore, most polypeptides in the cell require assistance for proper folding. A large class of proteins called molecular chaperones guide the folding process by binding to (or even encapsulating) polypeptides with hydrophobic exteriors, and through numerous rounds of ATP-fueled association and dissociation, facilitate proper folding and prevent aggregation. Chaperones also help maintain the structural integrity of mature proteins by binding to their exposed hydrophobic residues when they become misfolded, and thus have an important maintenance role in the cell. Chaperones are essential across all three domains of life (Bogumil et al. 2014), and in eukaryotes, organelles such as the endoplasmic reticulum (ER) and mitochondria have specialized chaperones.
But a paradox exists: Chaperones are themselves proteins, and thus require chaperones to be properly folded (fig. 11) (Boogerd et al. 2007, 238). This chaperones paradox poses a problem for the supposed naturalistic origin of chaperones and for the viability of early cells, which would have at some point lacked chaperones (Swee-eng 1996).
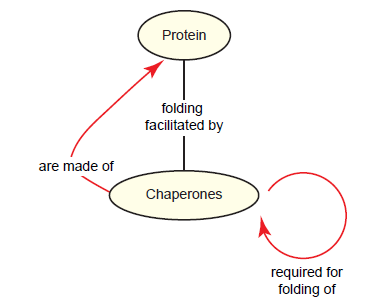
Fig. 11. The chaperones paradox. A team of chaperones mediates the folding of virtually all proteins in the cell. Since chaperones are themselves proteins, they require other chaperones for proper folding.
Naturally, evolutionists have acknowledged and responded to this issue. First, it can be pointed out that some chaperone folding can be facilitated by a different type of chaperone, such as appears to be the case with the E. coli chaperone GroEL, which assists in folding GroES (Moparthi et al. 2014). Even if this scenario is widespread, it does not skirt the issue that the earliest chaperone would have had no such assistance. The evolution of chaperones is addressed in the literature, but the most pithy rebuttal to this point comes from the blog of biochemist Larry Moran (Moran 2011):
In the beginning, you didn’t need chaperones because every protein folded rapidly on its own. Some of these primitive proteins might have been a bit slow to fold so the evolution of the first chaperones was advantageous because it enhanced the rate of folding for these proteins. The chaperones weren’t absolutely necessary for survival but they conferred a selective advantage on those cells that had them.
Once chaperones were present, new proteins could evolve that would otherwise have been too slow to fold in the absence of chaperones. Over time, cells accumulated more and more of these slowly folding proteins so that today no cell can survive without chaperones.
This is a plausible scenario if one readily accepts the evolutionary framework. Aside from that, it hinges on the fact that many polypeptides have the intrinsic ability to self-fold. This is certainly true in vitro (Hartl and Hayer-Hartl 2009), but the cell is a crowded environment whose cytosol contains 300–400 g/L of total protein (Hartl, Bracher, and Hayer-Hartl 2011), virtually ensuring the aggregation of misfolded proteins in the absence of chaperones. Therefore, it must be assumed that early cells were not crowded and allowed newly synthesized polypeptides the time and space to self-fold into their most favorable conformations. Based on this assumption, chaperones are proposed to have evolved very early before protein crowding was a problem (Hartl, Bracher, and Hayer-Hartl 2011), permitting chaperones to self-fold and enabling new chaperones and chaperone-dependent proteins to evolve. The existence of early chaperones would have been advantageous for the cell in that it constituted a type of self-defense mechanism against improperly folded proteins (Csermely et al. 2003).
But in the end, the assumptions that all early proteins could self-fold and that the early cell had very low protein concentration are not evidence-based and exist as theoretical concepts within the evolutionary framework. The chaperones paradox remains problematic for the evolution of the cell.
A Triad of Paradoxes on Membrane and Organelle Formation
Cell membranes are critical for life as they enable a multitude of cellular processes. All cells are enclosed by a plasma membrane, which is comprised of a phospholipid bilayer embedded with diverse proteins. The plasma membrane is selectively permeable and allows a cell to maintain very specific conditions on the inside, with most important molecules being moved in and out of the cell via membrane transport proteins. Embedded signaling proteins such as receptors also allow the cell to sense its environment and communicate with other cells. In eukaryotes, about half of the volume of the cell is taken up by membrane-enclosed compartments (organelles) with highly specialized functions and diverse membrane composition. Despite this diversity, most of the phospholipids in the cell are synthesized in the ER membrane and shuttled to other organelles via a direct connection with the ER or through transport vesicles. Mitochondria also exchange phospholipids with the endomembrane system through direct connection with the ER, both synthesizing specific phospholipids for the rest of the cell and receiving phospholipids for itself (Gohil and Greenberg 2009; Osman, Voelker, and Langer 2011).
Phospholipids can be added to a membrane (i.e. growth), and sections of one membrane can be pinched off to add to another membrane. However, no cell possesses the ability to synthesize a membrane from scratch (Alberts et al. 2015, 648, 689–691). Even obscure membranes that were once proposed to form de novo, such as the autophagosome membrane and the poxvirus membrane, are now known to be derived from ER-mitochondria contact sites and breakage of the ER membrane, respectively (Hamasaki, Shibutani, and Yoshimori 2013; Moss 2015). This presents a paradox: Membrane synthesis requires preexisting membrane (fig. 12A). This universal rule, the membrane formation paradox, is problematic for the early biogenesis and maintenance of cell membranes from a naturalistic perspective.
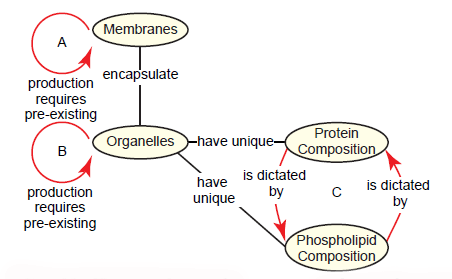
Fig. 12. The membrane formation paradox and its derivatives. A. The membrane formation paradox acknowledges that cell membranes can only be synthesized from pre-existing membranes. B. As an extension, the organelle formation paradox states that membrane-encapsulated organelles can only be produced from pre-existing organelles. C. The protein-phospholipid paradox demonstrates that protein composition in organelles is dictated by the organelle’s phospholipid composition, but the phospholipid composition is dictated by the protein composition.
If membrane can only be derived from preexisting membrane, how would early cells acquire a membrane? To this end, the fact that phospholipid bilayers can assemble spontaneously under the right conditions is comforting to many evolutionists. If dispersed into water, phospholipids will either form micelles or bilayers to avoid water interacting with their hydrophobic tails. And since even a bilayer sheet readily exposes many of these hydrophobic tails, the most energetically favorable conformation is a spherical bilayer, which can encapsulate materials (Alberts et al. 2015, 568–569). Most textbooks stop at this point as if to imply that the early evolution of the membrane would have been no big deal. But there are at least four major problems with this idea that are widely discussed in the literature.
First, the abiotic origin of amphipathic molecules (e.g. phospholipids) is problematic. Although chemists inspired by the Miller-Urey experiment have been able to synthesize a variety of organic molecules under very constrained conditions, early amphipathic molecules are widely believed to be of extraterrestrial origin, having originated on interstellar ices and transported to earth via meteorites (Deamer et al. 2002). These would have been less chemically complex than phospholipids, but could theoretically self-assemble into vesicular structures (Dworkin et al. 2001). This appeal to an extraterrestrial source for membrane-forming molecules is eerily similar to the proposed origin of homochirality in living systems, as discussed in a previous section. Even if this were true, it still makes the assumption that further terrestrial chemical evolution could produce complex phospholipids, which is wishful thinking.
The second problem with early membrane evolution is that Archaea and bacteria/eukaryotes possess very different membrane phospholipid composition and pathways for their biosynthesis. Cell membranes in bacteria and eukaryotes are comprised of glycerol-3-phosphate (G3P) ester-linked to fatty acid chains, whereas Archaea possess glycerol-1-phosphate (G1P) ether linked to isoprenoid chains. This striking difference between membranes and synthesis pathways in these groups have led many to suggest that early life did not have membranes at all, and that the two types of membranes evolved independently (Lombard, López-García, and Moreira 2012). However, even in this theoretical context, membrane encapsulation would have clear advantages such as protecting from parasitic RNA molecules and sequestering metabolites that would allow for the evolution of metabolism (Müller 2006). Therefore, various types of “proto-membranes” have been proposed for early life, such as those made of complex minerals or simple lipids synthesized without enzymes (Lombard, López-García, and Moreira 2012). Newer phylogenomic analyses, however, have rejected these proposals and led to the conclusion that cell membrane has existed throughout the entire evolution of life, and that the cenancestor had a modern-like membrane consisting of a mixture of both G1P and G3P phospholipids (Lombard, López-García, and Moreira 2012).
The third problem with early membrane evolution is that stable bilayer formation requires drastically different conditions than what is widely believed about the conditions under which early life evolved. From a chemistry perspective, the most reasonable scenario to spontaneously form stable membranes would be at temperatures below 60°C, with a near neutral pH and low ionic strength (Deamer et al. 2002; Müller 2006). This is in stark contrast to the accepted view of life originating near a hydrothermal vent in a marine environment.
Finally, early membrane evolution would have needed reliable mechanisms for growth, division, and nutrient transport. Since ready-made transport proteins and channels would have been absent from the hypothetical early membrane, such a membrane would be highly impermeable, thus defeating its purpose. Therefore, some have proposed a phospholipid chain length of 14 carbons instead of the 18-carbon length in modern day cells, which would increase its permeability by three orders of magnitude (Deamer et al. 2002). This membrane would be significantly thinner and highly unstable especially in the absence of embedded proteins, and still would not provide the selective permeability that is the hallmark of membranes in living cells. This seems to be mildly acknowledged since as one author puts it, “At some point early in evolution, a primitive transport system would have to evolve, perhaps in the form a polymeric compound that can penetrate the bilayer structure and provide a channel” (Deamer et al. 2002). Even if such a protein-free membrane could exist, growth and division would be a major problem since there would be nothing directing these processes. Naturally, speculation exists as to how this could occur. One idea—which assumes that early membranes were comprised of fatty acid/fatty acid alcohols—is that RNA replication would increase the osmotic pressure inside the “cell,” leading to growth by incorporating fatty acids from nearby membranes or micelles (Fry 2011; Müller 2006). Once the “cell” was large enough, shear force could split the membrane, forcing division (Müller 2006). Theories such as this hinge on the fact that some membrane breakages can spontaneously reassemble, but are otherwise playing with concepts of membrane biochemistry.
Since membranes encapsulate the vast majority of organelles, a natural extension of the membrane formation paradox is this: Organelle synthesis requires a pre-existing organelle (fig. 12B) (Nunnari and Walter 1996). We’ll call this the organelle formation paradox. The leading textbook in the field unpackages this clearly (Alberts et al. 2015, 648):
If the ER were completely removed from a cell, for example, how could the cell reconstruct it? . . . the membrane proteins that define the ER and perform many of its functions are themselves products of the ER. A new ER could not be made without an existing ER or, at least, a membrane that specifically contains the protein translocators required to import selected proteins into the ER from the cytosol (including the ER-specific translocators themselves). The same is true for mitochondria and plastids.
Thus, it seems that the information required to construct an organelle does not reside exclusively in the DNA that specifies the organelle’s proteins. Information in the form of at least one distinct protein that preexists in the organelle membrane is also required, and this information is passed from parent cell to daughter cells in the form of the organelle itself.
From this description, it is clear that the problems with organelle evolution go far beyond a consideration of the membrane. An organelle’s specific structure and function is largely due to its unique composition of phospholipids and proteins in its membrane. But what specifies the unique composition of each organelle? Phospholipid diversity and protein diversity in organelles are each dictated by the other. We’ll call this the protein-phospholipid paradox: Protein composition in an organelle is dictated by its phospholipid composition, which is in turn dictated by its protein composition (fig. 12C). The classic case of this is in the endomembrane system, which is comprised of the ER, Golgi, plasma membrane, and their byproducts. Although all membranes in this system are derived from the ER, each organelle (and even domains within an organelle) has a unique combination of phospholipids and proteins that are the basis for compartmental identity. As transport vesicles continuously bud from one compartment and fuse with another, molecular markers on the cytosolic face of the membranes ensure that fusion only occurs with the correct compartment. Both vesicle formation and fusion rely on proteins that recognize a specific type of phospholipid called phosphatidylinositol (PI), which can be phosphorylated in a number of unique ways. Each compartment has an exclusive combination of various phosphorylated forms of PI, thus ensuring that specific vesicle proteins assemble very selectively at each compartment. Interestingly though, the unique combinations of these lipids are determined by the sets of enzymes present in these membranes that alter PI and its derivatives. These enzymes are only localized specifically within compartments because of the specificity of vesicular transport regulated by the phospholipids in which they themselves act upon (Alberts et al. 2015, 697, 700–701, 710). This highlights an interdependent system that is all too common in the molecular biology of the cell.
Together, the three paradoxes discussed above pose significant problems for the evolution of membranes and organelles. As central as membrane biology is to the cell, especially in eukaryotes, it is clear that a slow, stepwise evolution of membranes would be a virtual miracle. But we have barely scratched the surface as to the complex processes that take place in organelle membranes. As an example of this, at last we will consider the paradoxical system regulating protein import into mitochondria.
The Mitochondrial Import Paradox
Mitochondria exist as a dynamic network of organelles that provide diverse and essential functions for the survival of eukaryotic cells. In addition to supplying energy for metabolism in the form of ATP, mitochondria participate in numerous biosynthesis pathways and play a central role in apoptosis. These and other processes require a massive number of proteins, and although mitochondria each contain a small genome, only 13 out of the ~1000 mitochondrial proteins in humans are made in-house, which are mostly specialized components of the respiratory chain (Alberts et al., 2015, 804). The overwhelming majority of proteins must be imported into mitochondria.
Mitochondrial proteins are first synthesized as precursors on cytosolic ribosomes and contain either cleavable or internal signal sequences that target them for mitochondrial import. Cytosolic chaperones prevent precursors from folding as they are directed to translocase channels on the outer surface of the mitochondrial membrane. Mitochondrial precursors first interact with the TOM (translocase of the outer membrane) complex (fig. 13). Although this process generally occurs post-translationally, there is mounting evidence for co-translational import for some proteins (Wenz et al. 2015). In either case, TOM serves as the entry gate, and depending on the type of protein and its intended destination, TOM sorts the precursor into a specialized sorting pathway (Wenz et al. 2015). In yeast—from which most data on this process has been obtained (Dimmer and Rapaport 2010)—α-helical outer membrane proteins are embedded by the MIM (mitochondrial import machinery) complex. β-barrel proteins are first translocated through TOM, then escorted by chaperones of the intermembrane space known as small TIMs to the SAM (sorting and assembly machinery) complex, where they are embedded in the outer membrane. Proteins with cysteine-rich motifs intended to remain in the intermembrane space are covalently modified by the MIA (mitochondrial intermembrane space import and assembly machinery) complex to prevent further translocation after emerging from TOM. Carrier proteins, after passing through TOM, are chaperoned by small TIMs to the TIM22 (translocase of the inner membrane) complex, and are embedded in the inner mitochondrial membrane. Single-pass inner membrane proteins are transferred directly to the TIM23 complex and embedded, whereas matrix proteins are translocated through TIM23 coupled with the PAM (presequence translocase-associated import motor) complex, using both ATP and membrane potential to drive translocation into the matrix (Alberts et al. 2015, 661–662; Fox 2012; Stojanovski et al. 2012; Wenz et al. 2015).
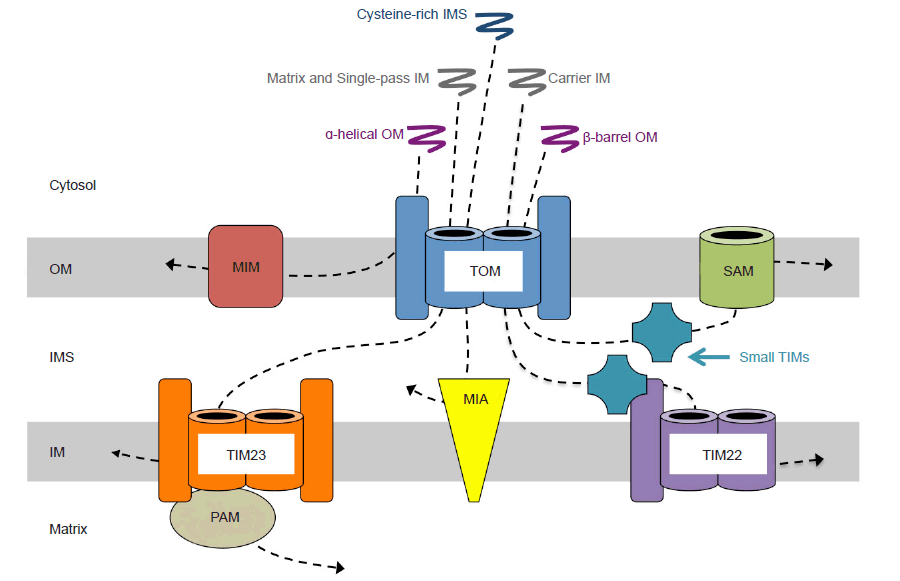
Fig. 13. Mitochondrial protein import routes. All imported mitochondrial precursor proteins first encounter the translocase of the outer membrane (TOM) complex, where they are then sorted to other import machinery. Alpha-helical outer membrane precursors are recognized by TOM, passed on to the mitochondrial import (MIM) complex, and embedded in the outer membrane. Matrix precursors and single-pass inner membrane precursors translocate through TOM and then the translocase of the inner membrane (TIM) 23 complex. Inner membrane proteins are embedded by TIM23, whereas matrix proteins are moved into the matrix with assistance from the presequence translocase-associated motor (PAM) complex. Cysteine-rich intermembrane space precusors pass through TOM and are modified by the intermembrane space import and assembly (MIA) complex to prevent translocation across the inner membrane. Carrier protein precursors pass through TOM and are guided by small TIM proteins to the TIM22 complex to be inserted into the inner membrane. -Precursors to beta-barrel proteins of the outer membrane translocate through TOM, and are guided by small TIMs to the sorting and assembly machinery (SAM), which facilitates embedding into the outer membrane. Abbreviations: OM, outer membrane; IMS, intermembrane space; IM, inner membrane.
The process of importing proteins into mitochondria is the most complex of any organelle in the cell and perhaps the most paradoxical. Mitochondrial import complexes are proteins themselves; therefore, they must be imported into mitochondria in the same manner as any other protein. And since they vary widely in composition and final destination within mitochondria, they rely upon intact import networks for their own localization. An extensive analysis of this has revealed a general theme, which we’ll call the mitochondrial import paradox: Import complexes require preexisting copies of themselves plus at least one other type of import complex in order to be properly localized into mitochondria (fig. 14). Many aspects of this paradox have been directly tested, while others are inferred based on the well-established model for mitochondrial protein import (Ahting et al. 2005; Becker et al. 2008; Dimmer et al. 2012; Dimmer and Rapaport 2010; Fox 2012; Papić et al. 2013; Stojanovski et al. 2012; Wenz et al. 2015). Subunitspecific details are listed in Table 1.
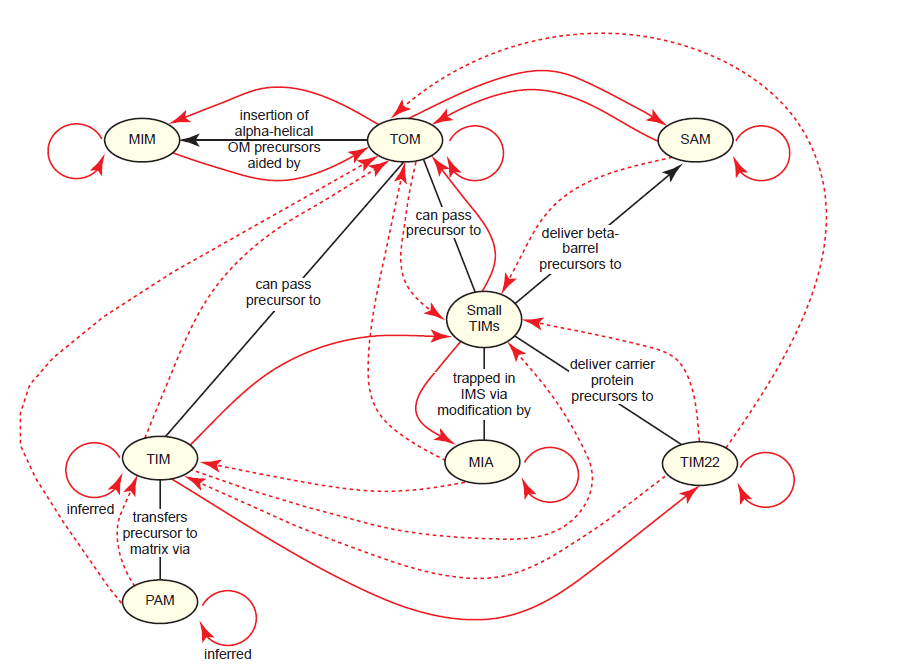
Fig. 14. The mitochondrial import paradox. All of the proteins comprising mitochondrial import complexes are synthesized in the cytosol, and therefore, must be imported into the mitochondria by pre-existing mitochondrial import complexes. Black lines highlight some basic interactions between components. Thick red lines indicate which import complexes regulate the import of the import complexes themselves, as shown with arrowheads. For example, every complex requires TOM since TOM serves as the gateway for virtually all imported proteins. Each solid red arrow indicates that at least one subunit of the import complex has been shown to be involved in the interaction described, mostly from data obtained in yeast; the specific subunits are omitted here for the sake of simplicity. Dashed arrows indicate that the requirement is inferred from models of mitochondrial protein import, but that direct data either does not yet exist or could not be located. The general theme is that most import complexes require pre-existing copies of themselves plus at least one other type of import complex in order to be properly localized into mitochondria.
| Complex or Subunit | Requires for Import | Comments | Reference(s) |
|---|---|---|---|
| Tom40 | Tom40; SAM complex; Mim1; Tim9-Tim10* | Tom40 is the β-barrel subunit of the TOM complex | Becker et al. 2008; Dimmer et al. 2012; Stojanovski et al. 2012; Wenz et al. 2015 |
| Tom22 | TOM complex; SAM complex | Tom22 is a receptor subunit of the TOM complex | Becker et al. 2008; Stojanovski et al. 2012 |
| Tom20 | Tom40; Mim1; Mim2 | Tom20 is a receptor subunit of the TOM complex | Ahting et al. 2005; Becker et al. 2008; Dimmer et al. 2012; Fox 2012 |
| Tom70 | Tom40; Mim1 | Tom70 is a receptor subunit of the TOM complex | Ahting et al. 2005; Becker et al. 2008; Fox 2012 |
| Mim1 | Mim2; Tom70; Mim1* | Mim1 is an α-helical OM protein | Dimmer et al. 2012; Dimmer and Rapaport 2010; Fox 2012; Papic et al. 2013; Wenz et al. 2015 |
| Sam50 | Sam50; Tom40; Tim9-Tim10* | Sam50 is the β-barrel subunit of the SAM complex | Fox 2012; Paschen, Neupert, and Rapaport 2005; Wenz et al. 2015 |
| Tim9-Tim10 | MIA complex | Tim9-Tim10 is an IMS chaperone complex | Fox 2012 |
| Tim13 | Tom22; Tom5 | Tim13 is an IMS chaperone | Fox 2012 |
| Erv1 | Mia40 | Erv1 is a subunit of the MIA complex | Fox 2012 |
| Mia40 | TOM complex*; TIM23 complex* | Mia40 is an IM-embedded subunit of the MIA complex | Wenz et al. 2015 |
| TIM23 | TOM complex* | Wenz et al. 2015 | |
| Tim17 | Tim22 | Tim17 is an essential subunit of the TIM23 complex | Rassow et al. 1999 |
| Tim23 | Tim8-Tim13; Tim22 | Tim23 is the channel subunit for the TIM23 complex | Paschen et al. 2000; Rassow et al. 1999 |
| Tim21 | TIM22 complex* | Tim21 is a subunit of the TIM23 complex | Wenz et al. 2015 |
| Tim50 | TIM22 complex* | Tim50 is a subunit of the TIM23 complex | Wenz et al. 2015 |
| mtHsp70 | TOM complex*; TIM23 complex*; mtHsp70* | mtHsp70 is a subunit of the PAM complex | Wenz et al. 2015 |
| TIM22 | Tim18 | Tim18 is a subunit of the TIM22 complex | Wenz et al. 2015 |
| Tim22 | TOM complex*; Tim9-Tim10*; Tim22 | Tim22 is the channel subunit of the TIM22 complex | Rassow et al. 1999; Wenz et al. 2015 |
| Tim54 | TOM complex*; TIM23 complex* | Tim54 is a receptor subunit of the TIM22 complex | Wenz et al. 2015 |
The mitochondrial import paradox presents a big problem for the evolution of mitochondrial protein import, and for the evolution of mitochondria in general. It seems obvious that such a complex system so vital to mitochondrial function could not evolve in a stepwise fashion, and that anything less than a fully intact import system would prevent entire classes of proteins (including import complexes themselves) from being embedded in mitochondria. But since the focus in the field is on how the system evolved and not whether it evolved (Lithgow and Schneider 2010), many evolutionists have embraced the challenge to explain the origin of the mitochondrial protein import machinery, and in doing so, attempt to demonstrate the grandeur of evolution.
Evolutionists almost universally accept the notion that mitochondria are derived from a double-membraned α-proteobacterium that was engulfed by another prokaryote and subsequently developed a symbiotic relationship (Lithgow and Schneider 2010). Even though bacteria do not import proteins and therefore lack protein import machinery, the mere presence of a protein-rich double membrane would at least, in theory, give evolution the raw materials to tinker with during the α-proteobacteria-to-mitochondria transition. Indeed, a mechanism for protein import would have been a necessary part of this transition since the vast majority of the α-proteobacterial genome would have (according to the model) been transferred to the nucleus. Hypotheses as to how the mitochondrial import machinery evolved are derived mainly from phylogenomic analyses. Surprisingly, the consensus is that even though mitochondria are supposedly of prokaryotic origin, only select subunits of the SAM and PAM complexes have orthologs in prokaryotes (Dolezal et al. 2006; Liu et al. 2011), none of which have any known function in protein transport (Clements et al. 2009). So even though the primitive α-proteobacterium would have theoretically had these orthologous proteins as starting material for the evolution of protein import, the implication is that the vast majority of subunits (~30 out of 35) would had to have been either created de novo or “recruited” from host cell genes (Clements et al. 2009; Hewitt, Lithgow, and Waller 2014; Liu et al. 2011). Moreover, since all eukaryotes share the same core machinery for mitochondrial protein import, this core machinery would have evolved quickly and early in eukaryotic evolution, before any lineage diversification. Other additions or deletions to the machinery, or the rewiring of import pathways, would have occurred later in a lineage-specific fashion (Dolezal et al. 2006; Hewitt, Gabriel, and Traven 2014; Hewitt, Lithgow, and Waller 2014; Liu et al. 2011).
Despite the extensive literature on the evolution of mitochondrial protein import pathways, the discussion has focused almost entirely on the order of events as informed by phylogenomic analyses. Several key questions have been largely ignored. First, how could the de novo creation of key import subunits, or the “recruitment” of other genes for use in import, mechanistically be achieved? Other than appealing to the seemingly magical power of random mutation, this question is scarcely addressed. One group, in a straw man rebuttal to Behe’s Irreducible Complexity, proposed a minimal set of events in which the TIM23 complex could have evolved by evolving binding interactions between preexisting bacterial machinery and converting an amino acid transporter to accept polypeptide chains (Clements et al. 2009). However, their frequent appeal to teleological language demonstrates that even the simplest of evolutionary scenarios requires a lot of wishful thinking (Luskin 2009).
Second, what drove the mitochondrial genome to be transferred to the nucleus during early eukaryotic evolution, thus necessitating a process for protein import in the first place? One paper appropriately frames this as a chicken-and-egg scenario (Dolezal et al. 2006):
It remains difficult to gauge the metabolic nature of the original symbiosis and, therefore, difficult to know what factors might have driven the ancient bacterial symbiont to surrender its genome and become a mere organelle of the host cell. Whatever the metabolic advantages, the majority of proteins functioning in mitochondria are now coded on nuclear genes. In a classic ‘chicken and egg’ scenario, it remains equally possible that the susceptibility of the endosymbiont to lose genes provided the selective pressure to create machinery to promote protein import, or that installing a protein import machinery enabled the productive transfer of endosymbiont genes to the nucleus.
Although evolutionists may construct an answer to this question at some point, it is clearly untestable, aside from being based on false premises.
Third, even if most of the mitochondrial genome was transferred to the nucleus, how could they acquire all of the complex regulatory sequences needed to allow their transcription, translation, and (important in this context) their targeting to a specific mitochondrial import pathway? The types of regulatory sequences that genes can possess are too numerous to list here, and even a perfectly functional mitochondrial protein import system is worthless unless proteins already contain the sequences necessary for mitochondrial import.
And finally, how could mitochondrial protein import evolve given that import proteins and respiratory chain proteins function interdependently? Interestingly, there is a fallout paradox here: Respiratory chain proteins are moved across the inner membrane by mitochondrial import proteins, utilizing the membrane potential generated by respiratory chain proteins (fig. 15) (Kulawiak et al. 2013). Recent research has illuminated the interconnectivity of mitochondrial protein import with cellular processes as diverse as metabolism, signaling, and stress, and that it is subject to intricate regulation (Harbauer et al. 2014). As our understanding of the mitochondrial protein interactome grows, it will likely present new issues for the purported evolution of mitochondria.
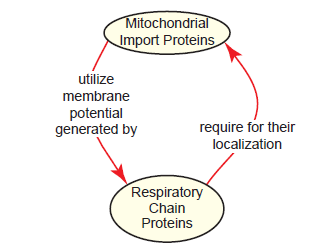
Fig. 15. The mitochondrial import-respiratory chain paradox. Mitochondrial import proteins and respiratory chain proteins are functionally interdependent. Respiratory chain proteins are moved across the mitochondrial inner membrane via import proteins, using the membrane potential generated by other respiratory chain proteins.
Conclusions
A major goal of this review was to demonstrate that the DNA-protein paradox remains as thorny as ever, and that it is only one of many paradoxes that plague cell and molecular biology from a naturalistic origins perspective. These nine “other” paradoxes span diverse cellular functions, and many are functionally interrelated, further highlighting the problems with naturally evolving these systems. Nevertheless, it is clear from the literature that evolutionists do not appear to be troubled by these paradoxes, as the truth of evolution is never questioned. Instead, entire fields of study have been built around studying the evolution of these systems, which further solidifies commitment to the theory, even in light of great complication. Many rebuttals to these paradoxes are ingenious, but are often based on theoretical constructs and are largely informed by phylogenomic analyses, which are only scientifically acceptable if one first assumes evolution. Interestingly, a frequent result of these phylogenomic analyses is that most of the critical components of a system evolved very early in evolution, with no observable or theoretically plausible intermediate forms. Very few evolutionary storylines even propose a stepwise evolution of system components, but instead rely on “magic words” (Guliuzza 2010) to make the evolutionary scenarios seem reasonable. In the end, what emerges is a fragmented storyline that fails to advance science or provide any ultimate satisfaction. But for the Christian, each of these paradoxes is instantly solved when one understands that all living systems were created intact by a wise and creative Creator.
Acknowledgments
Dr. Mike Gray and his teaching of Cell and Molecular Biology inspired a number of the ideas in this article. Dr. David Boyd critically reviewed the manuscript before submission and provided much support and helpful discussions. Courtney Cool Bonnema, a former student, did some initial research for the membrane paradox section. I also thank the Summer Institute in Teaching Science at Bob Jones University for providing financial support through the Science and Engineering Endowment Fund during the writing process. Inspiration® software was used to create the concept maps in the figures.
References
Ahting, U., T. Waizenegger, W. Neupert, and D. Rapaport. 2005. “Signal-anchored Proteins Follow a Unique Insertion Pathway into the Outer Membrane of Mitochondria.” Journal of Biological Chemistry 280 (1): 48–53.
Alberts, B., A. Johnson, J. Lewis, D. Morgan, M. Raff, K. Roberts, and P. Walter. 2015. Molecular Biology of the Cell. 6th ed. New York: Garland Science.
Aravind, L., D. R. Walker, and E. V. Koonin. 1999. “Conserved Domains in DNA Repair Proteins and Evolution of Repair Systems.” Nucleic Acids Research 27 (5): 1223–1242.
Babu, M. M., and S. A. Teichmann. 2003. “Evolution of Transcription Factors and the Gene Regulatory Network in Echerichia coli.” Nucleic Acids Research 31 (4): 1234–1244.
Becker, T., S. Pfannschmidt, B. Guiard, D. Stojanovski, D. Milenkovic, S. Kutik, N. Pfanner, C. Meisinger, and N. Wiedemann. 2008. “Biogenesis of the Mitochondrial TOM Complex: Mim1 Promotes Insertion and Assembly of Signal-anchored Receptors.” Journal of Biological Chemistry 283 (1): 120–127.
Blackmond, D. G. 2010. “The Origin of Biological Homochirality.” Cold Spring Harbor Perspectives in Biology 2 (5): 1–17.
Bogumil, D., D. Alvarez-Ponce, G. Landan, J. O. McInerney, and T. Dagan. 2014. “Integration of Two Ancestral Chaperone Systems into One: The Evolution of Eukaryotic Molecular Chaperones in Light of Eukaryogenesis.” Molecular Biology and Evolution 31 (2): 410–418.
Boogerd, F. C., F. J. Bruggeman, J. S. Hofmeyr, and H. V. Westerhoff, eds. 2007. Systems Biology: Philosophical Foundations. 1st ed. Oxford, United Kingdom: Elsevier.
Bourbon, H. M. 2008. “Comparative Genomics Supports a Deep Evolutionary Origin for the Large, Four-Module Transcriptional Mediator Complex.” Nucleic Acids Research 36 (12): 3993–4008.
Cheatle Jarvela, A. M., and V. F. Hinman. 2015. “Evolution of Transcription Factor Function as a Mechanism for Changing Metazoan Developmental Gene Regulatory Networks.” EvoDevo 6 (1): 1–11.
Ciccia, A., and S. J. Elledge. 2010. “The DNA Damage Response: Making It Safe to Play with Knives.” Molecular Cell 40 (2): 179–204.
Clements, A., D. Bursac, X. Gatsos, A. J. Perry, S. Civciristov, N. Celik, V. A. Likic, S. Poggio, C. Jacobs-Wagner, R. A. Strugnell, and T. Lithgow. 2009. “The Reducible Complexity of a Mitochondrial Molecular Machine.” Proceedings of the National Academy of Sciences of the United States of America 106 (37): 15791–15795.
Conaway, R. C., and J. W. Conaway. 2011. “Origins and Activity of the Mediator Complex.” Seminars in Cell and Developmental Biology 22 (7): 729–734.
Conova, S. 2003. “The Allure of Decorated Histones: Deciphering a Chemical Code That Controls Gene Expression.” In Vivo: The Newsletter of Columbia University Medical Center 2 (1). http://www.cumc.columbia.edu/publications/in-vivo/Vol2_Iss01_jan15_03/.
Crick, F. 1968. “The Origin of the Genetic Code.” Journal of Molecular Biology 38 (3): 367–379.
Csermely, P., C. Söti, E. Kalmar, E. Papp, P. Pato, A. Vermes, and A. S. Sreedhar. 2003. “Molecular Chaperones, Evolution and Medicine.” Journal of Molecular Structure (Theochem) 666–667: 373–380.
de Mendoza, A., A. Sebé-Pedrós, M.S. Šestak, M. Matejčić, G. Torruella, T. Domazet-Lošo, and I. Ruiz-Trillo. 2013. “Transcription Factor Evolution in Eukaryotes and the Assembly of the Regulatory Toolkit in Multicellular Lineages.” Proceedings of the National Academy of Sciences of the United States of America 110 (50): 1–9.
Deamer, D., J. P. Dworkin, S. A. Sandford, M. P. Bernstein, and L. J. Allamandola. 2002. “The First Cell Membranes.” Astrobiology 2 (4): 371–381.
Dimmer, K. S., D. Papić, B. Schumann, D. Sperl, K. Krumpe, D. M. Walther, and D. Rapaport. 2012. “A Crucial Role for Mim2 in the Biogenesis of Mitochondrial Outer Membrane Proteins.” Journal of Cell Science 125 (14): 3464–3473.
Dimmer, K. S., and D. Rapaport. 2010. “The Enigmatic Role of Mim1 in Mitochondrial Biogenesis.” European Journal of Cell Biology 89 (2–3): 212–215.
Dobson, C. M. 2004. “Principles of Protein Folding, Misfolding and Aggregation.” Seminars in Cell and Developmental Biology 15 (1): 3–16.
Dolezal, P., V. Likic, J. Tachezy, and T. Lithgow. 2006. “Evolution of the Molecular Machines for Protein Import into Mitochondria.” Science 313 (5785): 314–318.
Dworkin, J. P., D. W. Deamer, S. A. Sandford, and L. J. Allamandola. 2001. “Self-Assembling Amphiphilic Molecules: Synthesis in Simulated Interstellar/Precometary Ices.” Proceedings of the National Academy of Sciences of the United States of America 98 (3): 815–819.
Fox, T. D. 2012. “Mitochondrial Protein Synthesis, Import, and Assembly.” Genetics 192 (4): 1203–1234.
Fry, I. 2011. “The Role of Natural Selection in the Origin of Life.” Origins of Life and Evolution of Biospheres 41 (1): 3–16.
Gilbert, W. 1986. “Origin of Life: The RNA World.” Nature 319 (6055): 618–618.
Gohil, V. M., and M. L. Greenberg. 2009. “Mitochondrial Membrane Biogenesis: Phospholipids and Proteins Go Hand in Hand.” Journal of Cell Biology 184 (4): 469–472.
Guerrier-Takada, C., K. Gardiner, T. Marsh, N. Pace, and S. Altman. 1983. “The RNA Moiety of Ribonuclease P Is the Catalytic Subunit of the Enzyme.” Cell 35 (3 Pt 2): 849–857.
Guliuzza, R. 2010. “Unmasking Evolution’s Magic Words.” Acts & Facts 39 (3): 10–11.
Hamasaki, M., S. T. Shibutani, and T. Yoshimori. 2013. “Up-to-Date Membrane Biogenesis in the Autophagosome Formation.” Current Opinion in Cell Biology 25 (4): 455–460.
Hanawalt, P. C, and G. Spivak. 2008. “Transcription-Coupled DNA Repair: Two Decades of Progress and Surprises.” Nature Reviews Molecular Cell Biology 9 (12): 958–970.
Harbauer, A. B., R. P. Zahedi, A. Sickmann, N. Pfanner, and C. Meisinger. 2014. “The Protein Import Machinery of Mitochondria—A Regulatory Hub in Metabolism, Stress, and Disease.” Cell Metabolism 19 (3): 357–372.
Hartl, F. U., A. Bracher, and M. Hayer-Hartl. 2011. “Molecular Chaperones in Protein Folding and Proteostasis.” Nature 475 (7356): 324–332.
Hartl, F. U., and M. Hayer-Hartl. 2009. “Converging Concepts of Protein Folding in vitro and in vivo.” Nature Structural & Molecular Biology 16 (6): 574–581.
Hewitt, V. L., K. Gabriel, and A. Traven. 2014. “The Ins and Outs of the Intermembrane Space: Diverse Mechanisms and Evolutionary Rewiring of Mitochondrial Protein Import Routes.” Biochimica et Biophysica Acta 1840 (4): 1246–1253.
Hewitt, V., T. Lithgow, and R. F. Waller. 2014. “Modifications and Innovations in the Evolution of Mitochondrial Protein Import Pathways.” In Endosymbiosis. Edited by W. Loffelhardt, 19–36. Springer.
Higgs, P. G., and N. Lehman. 2015. “The RNA World: Molecular Cooperation at the Origins of Life.” Nature Reviews Genetics 16 (1): 7–17.
Ivica, N. A., B. Obermayer, G. W. Campbell, S. Rajamani, U. Gerland, and I. A. Chen. 2013. “The Paradox of Dual Roles in the RNA World: Resolving the Conflict Between Stable Folding and Templating Ability.” Journal of Molecular Evolution 77 (3): 55–63.
Joyce, G. F. 2002. “The Antiquity of RNA-Based Evolution.” Nature 418 (6894): 214–221.
Koonin, E. V. 2006. “DNA Repair: Evolution.” In Encyclopedia of Life Sciences, 1–4. Hoboken, New Jersey: John Wiley & Sons, Ltd.
Koster, M. J. E., B. Snel, and H. Th. M. Timmers. 2015. “Genesis of Chromatin and Transcription Dynamics in the Origin of Species.” Cell 161 (4): 724–736.
Kruger, K., P. J. Grabowski, A. J. Zaug, J. Sands, D. E. Gottschling, and T. R. Cech. 1982. “Self-Splicing RNA: Autoexcision and Autocyclization of the Ribosomal RNA Intervening sequence of Tetrahymena.” Cell 31 (1): 147–157.
Kulawiak, B., J. Höpker, M. Gebert, B. Guiard, N. Wiedemann, and N. Gebert. 2013. “The Mitochondrial Protein Import Machinery has Multiple Connections to the Respiratory Chain.” Biochimica et Biophysica Acta 1827 (5): 612–626.
Lindahl, T., and D. E. Barnes. 2000. “Repair of Endogenous DNA Damage.” Cold Spring Harbor Symposia on Quantitative Biology 65: 127–133.
Lithgow, T., and A. Schneider. 2010. “Evolution of Macromolecular Import Pathways in Mitochondria, Hydrogenosomes and Mitosomes.” Philosophical Transactions of the Royal Society of London, Series B, Biological Sciences 365 (1541): 799–817.
Liu, Z., X. Li, P. Zhao, J. Gui, W. Zheng, and Y. Zhang. 2011. “Tracing the Evolution of the Mitochondrial Protein Import Machinery.” Computational Biology and Chemistry 35 (6): 336–340.
Lombard, J., P. López-García, and D. Moreira. 2012. “The Early Evolution of Lipid Membranes and the Three Domains of Life.” Nature Reviews Microbiology 10 (7): 507–515.
Luskin, C. 2009. “PNAS Authors Resort to Teleological Language in Failed Attempt to Explain Evolution of Irreducible Complexity.” Evolution News and Views. http://www.evolutionnews.org/2009/09/pnas_knocks_down_straw_man024881.html.
Madhani, H. D. 2013. “The Frustrated Gene: Origins of Eukaryotic Gene Expression.” Cell 155 (4): 744–749.
Malik, H. S., and S. Henikoff. 2003. “Phylogenomics of the Nucleosome.” Nature Structural Biology 10 (11): 882–891.
Mariño-Ramírez, L., I. K. Jordan, and D. Landsman. 2006. “Multiple Independent Evolutionary Solutions to Core Histone Gene Regulation.” Genome Biology 7 (12): R122.
Marino, M. 2007. “Histone Code May Contain Secrets to Cell Differentiation.” Vanderbilt University Medical Center Reporter. http://www.mc.vanderbilt.edu:8080/reporter/index.html?ID=5669.
McCombs, C. 2014. “The Extraterrestrial Search for the Origin on Homochirality.” Creation Research Society Quarterly 51 (1): 5–13.
Moparthi, S. B., D. Sjölander, L. Villebeck, B. H. Jonsson, P. Hammarström, and U. Carlsson. 2014. “Transient Conformational Remodeling of Folding Proteins by GroES—Individually and in Concert with GroEL.” Journal of Chemical Biology 7 (1): 1–15.
Moran, L. A. 2011. “Protein Folding, Chaperones, and IDiots.” August 20, 2011. Retrieved June 9, 2016, from http://sandwalk.blogspot.com/2011/08/protein-folding-chaperones-and-idiots.html.
Moss, B. 2015. “Poxvirus Membrane Biogenesis.” Virology 479–480: 619–626.
Müller, U. F. 2006. “Re-creating an RNA World.” Cellular and Molecular Life Sciences 63 (11): 1278–1293.
Nunnari, J., and P. Walter. 1996. “Regulation of Organelle Biogenesis.” Cell 84 (3): 389–394.
O’Brien, P. J. 2006. “Catalytic Promiscuity and the Divergent Evolution of DNA Repair Enzymes.” Chemical Reviews 106 (2): 720–752.
Orgel, L. E. 1968. “Evolution of the Genetic Apparatus.” Journal of Molecular Biology 38 (3): 381–393.
Orgel, L. E. 2004. “Prebiotic Chemistry and the Origin of the RNA World.” Critical Reviews in Biochemistry and Molecular Biology 39 (2): 99–123.
Osman, C., D. R. Voelker, and T. Langer. 2011. “Making Heads or Tails of Phospholipids in Mitochondria.” Journal of Cell Biology 192 (1): 7–16.
Papić, D., Y. Elbaz-Alon, S. N. Koerdt, K. Leopold, D. Worm, M. Jung, M. Schuldiner, and D. Rapaport. 2013. “The Role of Djp1 in Import of the Mitochondrial Protein Mim1 Demonstrates Specificity Between a Cochaperone and its Substrate.” Molecular and Cellular Biology 33 (20): 4083–4094.
Paschen, S. A., W. Neupert, and D. Rapaport. 2005. “Biogenesis of Beta-Barrel Membrane Proteins of Mitochondria.” Trends in Biochemical Sciences 30 (10): 575–582.
Paschen, S. A., U. Rothbauer, K. Káldi, M. F. Bauer, W. Neupert, and M. Brunner. 2000. “The Role of the TIM8-13 Complex in the Import of Tim23 into Mitochondria.” The EMBO Journal 19 (23): 6392–6400.
Pusarla, R., and P. Bhargava. 2005. “Histones in Functional Diversification: Core Histone Variants.” The FEBS Journal 272 (20): 5149–5168.
Rassow, J., P. J. Dekker, S. van Wilpe, M. Meijer, and J. Soll. 1999. “The Preprotein Translocase of the Mitochondrial Inner Membrane: Function and Evolution.” Journal of Molecular Biology 286 (1): 105–120.
Robertson, M. P., and G. F. Joyce. 2012. “The Origins of the RNA World.” Cold Spring Harbor Perspectives in Biology 4 (5): 1–22.
Sarfati, J. 2000. “Origin of Life and the Homochirality Problem: Is Magnetochiral Dichroism the Solution?” Journal of Creation 14 (3): 9–12.
Stojanovski, D., M. Bohnert, N. Pfanner, and M. van der Laan. 2012. “Mechanisms of Protein Sorting in Mitochondria.” Cold Spring Harbor Perspectives in Biology 4 (10): a011320.
Swee-eng, A. W. 1996. “The Origin of Life: A Critique of Current Scientific Models.” Creation Ex Nihilo Technical Journal 10 (3): 300–314.
Turner, B. M. 2014. “Nucleosome Signalling; An Evolving Concept.” Biochimica et Biophysica Acta 1839 (8): 623–626.
Visweswariah, S. S., and S. J. W. Busby. 2015. “Evolution of Bacterial Transcription Factors: How Proteins Take on New Tasks, But Do Not Always Stop Doing the Old Ones.” Trends in Microbiology 23 (8): 463–467.
Wenz, L-S, Ł. Opaliński, N. Wiedemann, and T. Becker. 2015. “Cooperation of Protein Machineries in Mitochondrial Protein Sorting.” Biochimica et Biophysica Acta 1853 (5): 1119–1129.
Woese, C. R. 1967. The Genetic Code: The Molecular Basis for Genetic Expression. New York: Harper & Row.
Yin, J., and G. Wang. 2014. “The Mediator Complex: A Master Coordinator of Transcription and Cell Lineage Development.” Development 141 (5): 977–987.













