The views expressed in this paper are those of the writer(s) and are not necessarily those of the ARJ Editor or Answers in Genesis.
Abstract
All known organisms, except RNA viruses, contain a DNA genome. The percentage of the part of the genome encoding genes, including protein-coding and non-protein coding genes, differ greatly among organisms. Regardless, all genes are encoded in DNA or RNA. In all cells, DNA and RNA are generated using DNA or RNA templates. This article argues, based on theoretical considerations, as well as bottom-up and top-down experimental data, that it is practically impossible to generate the first DNA (or RNA) template abiotically.
Keywords: abiogenesis, origin of life, chemical evolution, self-organization, RNA world
Introduction
“Anybody who thinks they know the solution to this problem (of the origins of life) is deluded,” said the Salk Institute Leslie Orgel, a pioneer and an impeccable and quintessential practitioner of prebiotic chemistry (Bardi 2004).
“Understanding the chemical origins of DNA and RNA in the context of origins of life continues to be an enigma,” stated the Scripps Research Institute Ramanarayanan Krishnamurthy, the renowned origin-of-life scientist who likes ending his talks about the origin-of-life by quoting Orgel’s above remark (Krishnamurthy 2018).
“The origin of genetic polymers remains a poorly understood phenomenon, but not from lack of investigation,” proclaimed Nicholas Hud, another prominent origin-of-life scientist, and his colleagues (Fialho et al. 2021).
Krishnamurthy further observed that various investigations of plausible pre-biotic pathways of generating DNA and RNA are “rooted in deep convictions—partly based on scientific facts and the remaining filled with extrapolated scientific imaginations about the early earth scenarios and the availability of source materials that could/would lead to the building blocks of, and eventually to, DNA and RNA” (Krishnamurthy 2018). He stated that the “wide-ranging solutions are under very different conditions that do not, at first or even second glance, seem to be compatible with one another.” Our lack of firm knowledge of the early earth conditions “permits each experimental approach to pick early Earth conditions tailored to its demonstrated synthetic pathways and stake the claim of relevance. One certainly can propose them, but only with the appreciation that they are broad approximations that can, and will, be challenged before ‘the ink is dry’ on the publication.”
Realizing the difficulties of abiotic generation of DNA and RNA templates, some propose a pre-RNA world that was made of proto-nucleotides. However, this creates more problems, including what were the proto-nucleotides, how they were generated abiotically, how they self-linked to form a pre-RNA world, and how and why this was changed into an RNA world. For example, Nicholas Hud and colleagues proposed “a new class of plausibly prebiotic polymer, depsipeptide nucleic acid (DepsiPNA), for which the monomers are nucleobase-functionalized α-hydroxy acid-α-amino acid heterodimers. These monomers are (1) consistent with water-soluble organic compounds believed to have been present on the early earth, (2) able to polymerize in model prebiotic reactions that require few special conditions, and (3) able to undergo complementary self-assembly and selective degradation” (Fialho et al. 2021). However, their proto-nucleotide-generating intermediates have to be synthesized under different “prebiotic” conditions. Their favorite melamine-functionalized hydroxy acid has to be synthesized in two steps, the first step needs an acidic solution (pH = 4) and the second step needs a basic (pH = 10) solution, a drop of proton concentration from 10-4 to 10-10 (Figure 3 of (Fialho et al. 2021)). The nucleobase-functionalized α-hydroxy acid interacts with L-aspartic acid to generate their proto-nucleotide monomers—nucleobase-functionalized α-hydroxy acid-α-amino acid heterodimers. The proposed oligomerization occurs at pH 2 (Fialho et al. 2021, Figure 5a), an increase of proton concentration from 10-10 to 10-2. It would be very difficult for the “prebiotic earth” to adjust its pH values to meet the requirements of different experimental steps in time, without intelligent input, to make these “proto-nucleotides” and their short (a few nucleotides long) oligomers.
In this essay, I invite you to consider the challenges in generating the first gene-encoding DNA or RNA genome abiotically, first from a theoretical point of view and then from experimental results gathered from literature. All theoretical considerations and experimental evidence suggest that a natural way of generating gene-encoding DNA or RNA, that is, DNA or RNA compositional monomers self-link to generate DNA and RNA, is practically impossible. Even if it did happen, self-linking monomers will destroy genetic information coding and decoding systems, as I described in “Facts Cannot be Ignored When Considering the Origin of Life #1” (Tan 2022a).
Theoretical Consideration
Calculating the possibilities of making a DNA genome with two basepairs
Assuming the top strand of our imaginary genome is a dideoxynucleotide with the sequence of 5’-AC-3’, a dinucleotide made of dAMP (deoxyadenosine-5’-monophosphate) and dCMP (deoxycytidine-5’-monophosphate). Since the bottom strand is the reverse and complementary of the top strand, its sequence must be 5’-GT-3’, a dideoxynucleotide made of dGMP (deoxyguanosine-5’-monophosphate) and dTMP (deoxythymidine-5’-monophosphate).
The number of possible isomers of our two-base-pair genome can be calculated in three steps: calculating the isomers of the first strand of the genomic DNA (5’-AC-3’), calculating the isomers of the second strand of DNA (5’-GT-3’) given the sequence of the first strand, and calculating the total isomers of both strands, as detailed below.
Calculating the number of isomers of the first strand of the genomic DNA
To calculate the number of possible isomers for the first strand of DNA of our imaginary genome, 5’-AC-3’, we need to determine the number of isomers of dAMP, the number of isomers of dCMP, and the possible ways of linking them together.
The number of isomers of one dAMP molecule can be calculated by multiplying the number of OH groups in a D-2-deoxyribose by the number of amine (NH) groups in an adenine that can be used to form carbon-nitrogen (C-N) bonds and by the number of possible ways of linking the resultant molecules to a phosphate group.
Note that D-2-deoxyribose exists in five forms in water solutions, each form has three OH groups (fig. 1) that can be used to link with an adenine via a C-N bond. The six-membered ring forms (fig. 1A and B) are the most stable, while the linear form (fig. 1C) is the least stable. It is interesting that the isomer used in DNA made by DNA polymerases is the five-membered ring form (fig. 1D and E) and only its ß form (ß-D-2-deoxyribofuranose, fig. 1E), instead of the more stable six-membered ring forms of D-2-deoxyribose. Note that even though the linear D-2-deoxyribose is present in only 0.7% in the equilibrium of an aqueous ribose solution at neutral pH, that does not mean that this form would only have 0.7% of opportunities to participate in a reaction because the stability of a chemical and its reactivity are two separate issues. Therefore, to simplify the calculations, each OH in the ribose ring is treated equally.
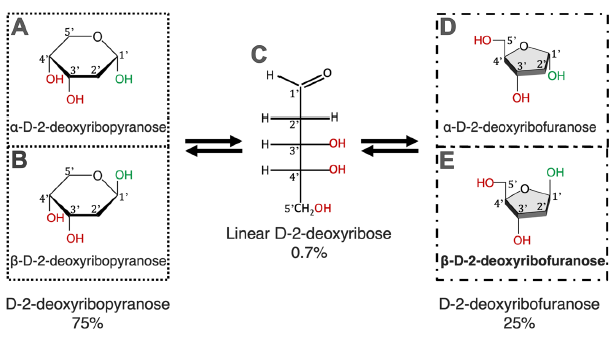
Fig. 1. Chemical equilibrium of D-2-deoxyribose in water. D-2-deoxyribose exists in five forms. All five forms exist in an equilibrium; none of which can exist alone. Percentages of different forms of D-2-deoxyribose are according to Akane700 for ja:Wikipedia (https://commons.wikimedia.org/w/index.php?curid=3267188).
Adenine exists in two forms in water solutions, thus there are three NH groups in an adenine molecule (N7H, N9H, and NH of the NH2 of C6) that can be used to link with the sugar ring via a C-N bond (fig. 2A). The number of NH groups that can be used to link to the sugar ring via a C-N bond in a cytosine is also three (N1H, N3H, and NH of the NH2 of C4) (fig. 2 B). That number is five for guanine (N1H, N3H, N7H, N9H, and NH of the NH2 of C2) and two for thymine or uracil (N1H and N3H) (Fig. 2 C–E). The two hydrogen atoms of the NH2 of C6 of adenine are indistinguishable and are counted as one NH group; the same is true for the NH2 of C4 of cytosine and the NH2 of C2 of guanine.
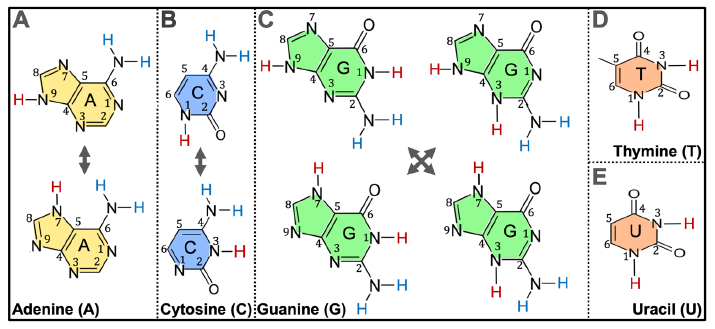
Fig. 2. Chemical equilibrium of nitrogenous bases in aqueous solutions. (A) Adenine. (B) Cytosine. (C) Guanine. (D) Thymine. (E) Uracil. The different forms of each base differ in the positions of the double bonds in their rings and thus the positions of the hydrogen atoms (red) that directly connect to the nitrogen atoms that are part of the rings. The two amine hydrogen atoms (blue) of adenine C6 are indistinguishable and are thus counted as one NH, so are those of cytosine C4 and of guanine C2.
Therefore, there are 3 (OH groups for each D-2-deoxyribose form) × 3 (NH groups in an adenine) × 5 (forms of D-2-deoxyribose) = 45 ways of linking an adenine with a D-2-deoxyribose via a C-N bond. The same is true for a cytosine. For a guanine molecule, there are 75 ways to link it with a D-2-deoxyribose via a C-N bond. Thymine and uracil both have 30 such ways.
For each particular D-2-deoxyribose form, when one of its three OH groups has been used to link adenine (or any other bases), there will be two OH groups left to be used to link to a phosphate group. Therefore, there are a total of 2 × 45 = 90 ways of linking an adenine, a D-2-deoxyribose, and a phosphate group together (fig. 3). That number is 90, 150, and 60 for cytosine, guanine, and thymine, respectively. These are the numbers of isomers of dNMP in which N is an adenine (A), a cytosine (C), a guanine (G), or a thymine (T), excluding all the isomers of D-2-deoxyribose that are not a D-2-deoxyribose, all the isomers of A/C/G/T that are not A/C/G/T and all the isomers of linking the phosphate group through non-phosphoester bonds.
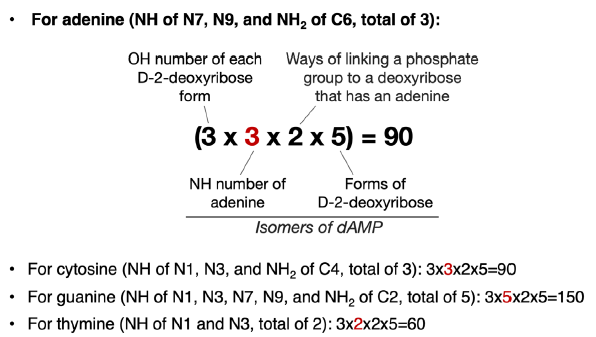
Fig. 3. Calculation of possible ways of joining a nitrogenous base, a phosphate group, and a D-2-deoxyribose. The number (highlighted in red) that varies in each equation is the number of NH groups of different bases that can be used to form C-N bonds with D-2-deoxyribose.
The last step of calculating the isomers of 5’-AC-3’ is to determine the possible ways of linking the isomers of dAMP and the isomers of dCMP together. In each of the isomers of dAMP or dCMP, one of the three OH groups of its D-2-deoxyribose is occupied by a nitrogenous base, another by a phosphate group. Therefore, only one OH left in each isomer of dAMP can be used to link to the phosphate group of an isomer of dCMP, and vice-versa. Consequently, there are two ways to link an isomer of dAMP and an isomer of dCMP. For example, a dAMP and a dCMP can be linked via a phosphodiester bond to form 5’-AC-3’, or 5’-CA-3’.
In reality, when one performs a chemical reaction, one would never use just one, but many, molecules of each reagent. Therefore, in a dehydration reaction to link A and C together, there are many As and Cs in the reaction mixture. Consequently, in addition to A and C linking together, A would also link with A and C would also link with C. Furthermore, the phosphate groups would also link to each other to form a pyrophosphate bond. It was found that, in a dehydration reaction of nucleotides, the formation of 5′,5′-pyrophosphate linkage is more favorable than the formation of 2′,5′-phosphodiester linkage, which is more favorable than the formation of the desired 3′,5′-phosphodiester linkage (see (Robertson and Joyce 2012) and also data discussed below). Indeed, it is hard to estimate how many undesired linkages would occur when one tries to link two deoxynucleotides together. This is a reflection of the Paradox of Asphalt or Tar of abiogenesis, which states: “An enormous amount of empirical data have established, as a rule, that organic systems, given energy and left to themselves, devolve to give uselessly complex mixtures, ‘asphalts’”(Benner 2014). To simplify our calculation of isomers of making a dinucleotide, we will just stay with two ways, both of which lead to the formation of phosphodiester bonds, but with opposite sequence (e.g., AC vs CA).
Therefore, there are 90 × 90 × 2 = 16,200 ways of linking an adenine, a cytosine, two D-2-deoxyribose, and two phosphate groups together via C-N bonds and phosphoester bonds (fig. 4).
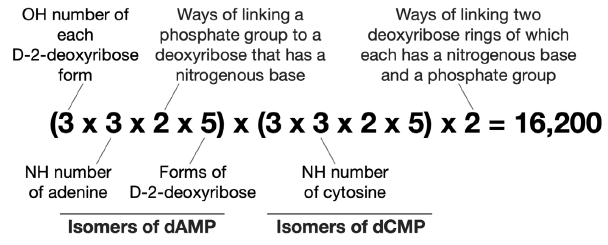
Fig. 4. Calculation of possible number of isomers of adenine cytosine D-2-deoxyribose dinucleotide.
It is worth pointing out that this calculation excludes all the isomers of which the atoms of D-2-deoxyribose are arranged to form something other than D-2-deoxyribose, or the atoms in adenine arranged to form something other than adenine, or the atoms in cytosine arranged to form something other than cytosine, or sugar rings not linked with the phosphate group through a phosphoester bond or a phosphodiester bond. The number of the probable nucleotide isomers that are excluded is much greater than the number of those that are included.
It is amazing that our desired adenine-cytosine D-2-deoxyribose dinucleotide is just one of the 16,200 possible chemicals that could result from linking an adenine, a cytosine, two D-2-deoxyribose, and two phosphate groups. Many of the isomers with the wrong configurations are energy-equivalent to or even more stable than the correct one, and some of the other forms would be detrimental to life.
Calculating the number of isomers of the second strand
As shown in Fig. 2, there are five NH groups that can be used to form C-N bonds with D-2-deoxyribose in guanine and two in thymine. Following the procedure for calculating the isomers of 5’-AC-3’, we get the number of isomers of 5’-GT-3’ as 18,000 (fig. 5).
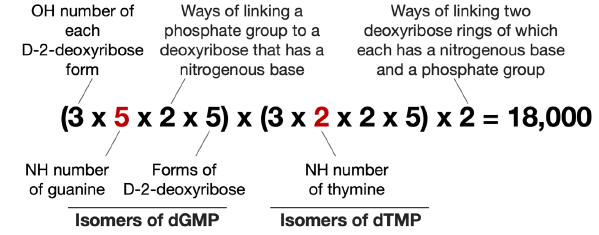
Fig. 5. Calculation of possible number of isomers of guanine-thymine D-2-deoxyribose dinucleotide. The differences of the calculation for the number of isomers of guanine-thymine D-2-deoxyribose dinucleotide and that of the adeninecytosine D-2-deoxyribose dinucleotide are highlighted in red.
However, the order of the nucleotides in the second strand is not independent. In a normal double-stranded DNA, the sequence of the second (complementary) strand of DNA is determined by the sequence of the first strand. If an isomer of A would pair and only pair with an isomer of T, and an isomer of G would pair and only pair with an isomer of C, there would be only one way of linking the nucleotides in the second strand. Therefore, the number of isomers of the second strand of our desired genome is equal to the number of isomers of dGMP × the number of isomers of dTMP × 1 = 9,000. This is a situation that is not strictly realistic because many of the isomers will have difficulty forming the hydrogen bonds that are needed for base pairing of the two strands of DNA isomers, or at least the bonds will not form as efficiently as the bonds between the normal nucleotides do. Therefore, the number of isomers of the second strand given is a minimum number.
Calculating the total number of isomers of the genome
The total isomer number of both strands of our desired genome is the product of the number of isomers of the first strand and that of the second strand: the number of isomers of the first strand (AC) X the number of isomers of the second strand (GT) = 16200 × 9000 = 145,800,000.
145,800,000 possible isomers for a DNA genome of only two basepairs!
Calculating the possibilities of making an RNA genome with two basepairs
For an easy comparison with the above situation of a double-stranded DNA genome, let’s assume that our double-stranded RNA genome contains the same nucleobases as our two basepair DNA genome, except changing thymine to uracil. Thus, the top strand of our imaginary RNA genome would be 5’-AC-3’, a dinucleotide made of AMP (adenosine-5’-monophosphate) and CMP (cytidine-5’-monophosphate), and the bottom strand would be 5’-GU-3’, a dinucleotide made of GMP (guanosine-5’-monophosphate) and UMP (uracil-5’-monophosphate).
Note that in place of the D-2-deoxyribose sugar in DNA, we have a D-ribose (in short ribose) sugar in RNA. As D-2-deoxyribose, ribose also exists in five configurations in water, whichever isoform is started with. What is different is that ribose has one additional OH group than D-2-deoxyribose (fig. 6).
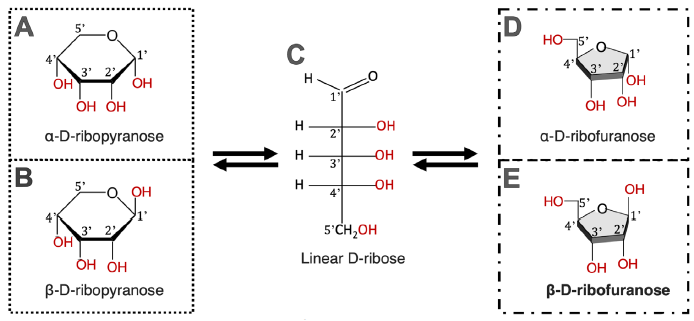
Fig. 6. Chemical equilibrium of D-ribose in water. Ribose exists in five forms. All five forms exist in an equilibrium; none of which can exist alone.
Calculating the number of isomers of the first strand of the double-stranded RNA
The change from D-2-deoxyribose to ribose not only increases the number of OH groups available to link with a nitrogenous base and a phosphate group, but also the ways of linking two nucleotides. Consequently, there are 4 (OH groups for each ribose form) × 3 (NH groups in an adenine) × 5 (forms of ribose) = 60 ways of linking an adenine with a ribose via a C-N bond. The same is true for a cytosine. For a guanine molecule, there are 100 (= 4 × 5 × 5) ways to link it with a ribose via a C-N bond. Thymine and uracil both have 40 (= 4 × 2 × 5) such ways.
For each particular ribose form, when one of its four OH groups has been used to link adenine (or any other bases), there will be three OH groups left to be used to link to a phosphate group. Therefore, there are a total of 3 × 60 = 180 ways of linking an adenine, a ribose, and a phosphate group together (fig. 7). That number is 180, 300, and 120 for cytosine, guanine, and uracil, respectively. These are the numbers of isomers of NMP in which N is an adenine (A), a cytosine (C), a guanine (G), or a uracil (U), excluding all the isomers of ribose that are not a ribose, all the isomers of A/C/G/U that are not A/C/G/U and all the isomers of linking the phosphate group through non-phosphoester bonds.
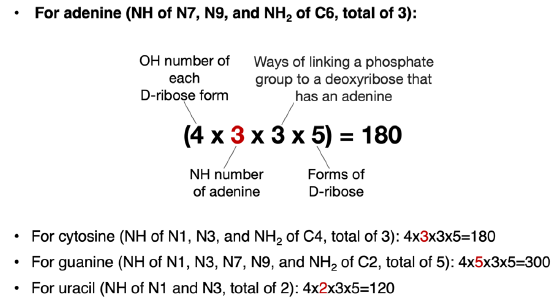
Fig. 7. Calculation of possible ways of joining a nitrogenous base, a phosphate group, and a ribose. The number (highlighted in red) that varies in each equation is the number of NH groups of different bases that can be used to form C-N bonds with ribose.
There are two ways of linking two deoxyribonucleotides, but there are four ways of linking two ribonucleotides for the following reasons. In each of the isomers of AMP or CMP, one of the four OH groups of its ribose is occupied by a nitrogenous base, another by a phosphate group. Therefore, there are two OH left in each isomer of AMP that can be used to link to the phosphate group of an isomer of CMP, and vice-versa. Consequently, there are four ways to link an isomer of AMP and an isomer of CMP. Taken together, an adenine-cytosine ribose dinucleotide has 129,600 possible configurations (fig. 8). In comparison, an AC di-deoxyribonucleotide has only 16,200 possible configurations. Consequently, it is even more difficult at every step to generate a correct RNA sequence than it is to generate DNA, given all the necessary nucleobases, sugar rings, and phosphate groups.
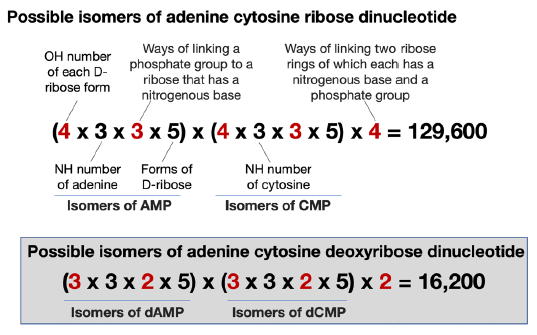
Fig. 8. A comparison of the calculation of isomers of 5’-AC-3’ RNA and DNA dinucleotides. The numbers differ between RNA and DNA are highlighted in red.
Calculating the number of isomers of the second RNA strand
Following the above procedure, we find that a single-stranded GU dinucleotide RNA has 144,000 possible isomers, comparing the 18,000 possible isomers of a GT di-deoxyribonucleotide (fig. 9). As with the case of double-stranded DNA, the sequence of the second strand of a double-stranded RNA is not independent. Thus, the isomers of GU, as the second strand of the double-stranded RNA, would be 72,000 (=144,000/2).
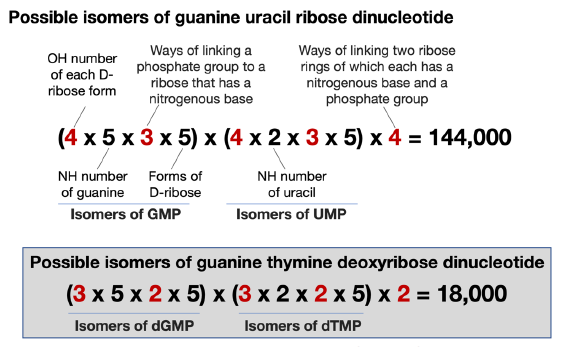
Fig. 9. A comparison of the calculation of isomers of a 5’-GU-3’ RNA dinucleotide and a 5’-GT-3’ DNA dinucleotide. The numbers differ in RNA and DNA are highlighted in red.
Calculating the total number of isomers of the double-stranded RNA genome
The total isomer number of both strands of our desired RNA genome is the product of the number of isomers of the first strand and that of the second strand: the number of isomers of the first strand (AC) X the number of isomers of the second strand (GU) = 129,600 × 72,000 = 9,331,200,000. This is 64 times the isomer number (145,800,000) of a double stranded AC/GT DNA.
Lessons from Experiments
Lessons from bottom-up studies
Untemplated uncatalyzed oligonucleotides formation
Free natural nucleotides, including AMP, ADP, and ATP, do not link themselves together to form polynucleotides or oligonucleotides (oligos) in water solutions. Artificially 5’-hydroxyl group-activated non-natural nucleotides, such as 5’-phosphorimidazolide and 5’-phosphoro-1-methyladenine of nucleosides (ImpN and MeadpN, N is any nitrogenous base) (fig. 10), could form a small amount of dimers and a trace amount of oligomers (Ertem and Ferris 1997; Ferris and Ertem 1993; Kawamura and Ferris 1999; Sulston et al. 1968; Weimann et al. 1968).
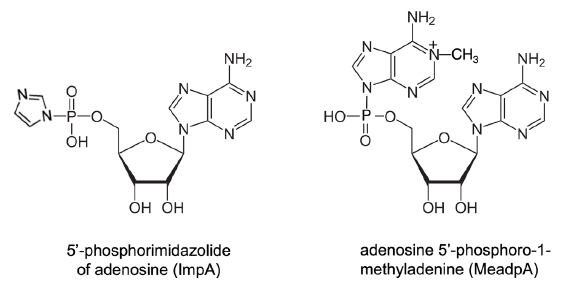
Fig. 10. Monomers of nucleotides with 5’ hydroxyl group selectively activated. The nucleobase (nitrogenous base) in the examples is adenine. The activated nucleotides need to be synthesized and stored in anhydrous conditions.
Note that the products of the reaction are much more complicated than what we have considered in our theoretical calculations (fig. 11). A major process is the hydrolysis of the activated starting monomer. Furthermore, a dimer or polymer can cyclize. The activated monomer also interacts with chemicals (e.g., HEPES) that are used to make the reaction buffer solutions. Consequently, the main products in such a reaction are 5’-phosphonucleotide (NMP, a hydrolysis product of the activated monomer), 5’,5’-pyrophosphate-, 2’,5’-or 3’,5’-phosphodiester bond-linked dimers, cyclic dimers, and a small percentage of longer oligomers (Kanavarioti 1997, 1998). In other words, most of the products are not the desired RNA polymers.
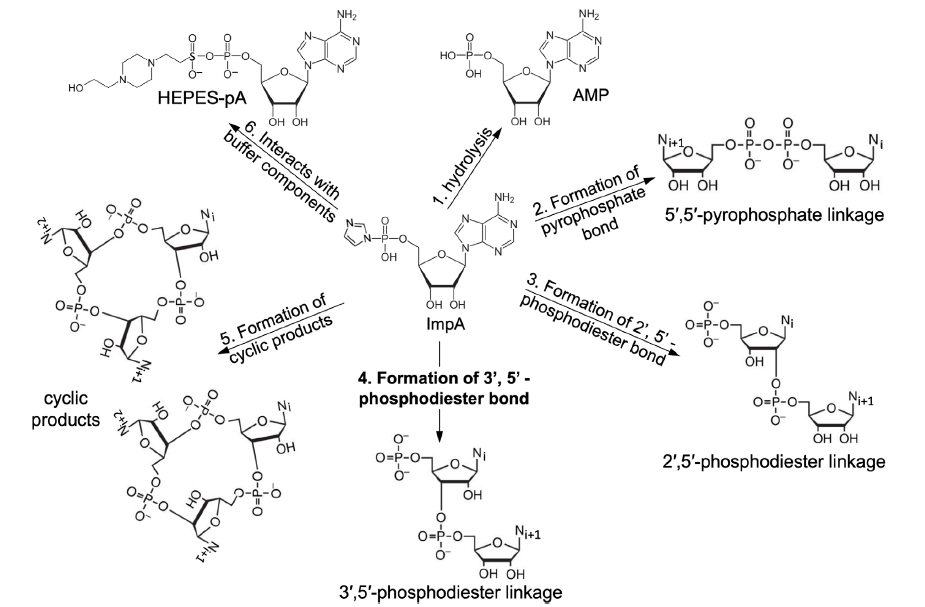
Fig. 11. Main reactions of activated mononucleotides in water. The nucleobase in this figure is adenine, which is represented with Ni, Ni+1, or Ni+2 in reactions 2-5. The reaction forming the desired 3’, 5’-phosphodiester bonds (that is, reaction 4) is bolded.
Increasing the concentration (up to 1.4 M) of the activated nucleotides has little effect on changing the length of the oligomers. Some metal ions and minerals can lengthen the generated oligomers (Ding, Kawamura, and Ferris 1996; Ferris and Ertem 1992, 1993; Kumar 2016; Sawai 1976, 1988; Sawai, Higa, and Kuroda 1992; Sawai, Kuroda, and Hojo 1989). The ratio of 3’,5’ vs 2’,5’ phosphodiester bonds in the products varies depending on the kinds and concentrations of ions present as well as the activation group of the nucleotides (Kumar 2016; Sawai 1976, 1988; Sawai, Higa, and Kuroda 1992; Sawai, Kuroda, and Hojo 1989).
Untemplated non-enzymatic oligos formation
No non-enzyme-based condition has been found to produce polynucleotides that are comparable with natural DNA and RNA in length, composition, manners of linkage between nucleotide monomers, and ways how the sequence of monomers in a specific DNA or RNA polymer is determined. Natural DNA and RNA molecules are made of four nucleobases (adenine, guanine, cytosine, and thymine for DNA or uridine for RNA) linked to D-ribose for RNA and D-2-deoxyribose sugars for DNA. The nucleotides are linked via, and only via, 3’,5’ phosphodiester bonds. The sequences of DNA and RNA in cells are determined by the sequences of their template DNA and the protein machineries replicating or transcribing the DNA instead of by the chemical or physical affinities of their constructing nucleotides.
Using Na+ montmorillonite clay as a catalyst
The most popular non-enzymatic method of generating oligos in origin-of-life studies is catalyzed oligomerization of activated nucleotides, including ImpN, MeadpN, and 5’-phosphate 2-methylimidazolide nucleosides (2-MeImpN). The most promising catalyst is Na+ montmorillonite clay. In its presence, oligomers containing 40–50 monomers can be generated from MeadpA and MeadpU, 20–25-mers from MeadpC, and oligoGs of unidentified length from MeadpG (Huang and Ferris 2006, 2003; Prabahar and Ferris 1997).
Using ImpN, a compound simpler and more widely used than MeadpA, oligos of around 14 nucleotides (nt) long (or 14mers) have been synthesized in one-pot reactions (Ertem and Ferris 1997; Ferris and Ertem 1993; Kawamura and Ferris 1999). Oligoadenylates (oligoA) of 40–50 nt and oligouridine (oligoU) of 25–30 nt were produced by elongation of a 10 nt oligoA, 32pdA(pdA)8pA, with the daily removal of spent reaction supernatants and addition of activated nucleotides, ImpA and ImpU (5’-phosphorimidazolide uridine), respectively, for up to 14 days (Ferris 2002; Ferris et al. 1996). Longer reaction times did not result in longer oligomers (Ferris 2002).
The formed oligomers decomposed via hydrolysis in the same reaction buffer lacking ImpA or ImpU, and the clay accelerated the hydrolysis.
A Goldilocks reaction condition is necessary for the elongation of the oligoA primer (Ferris 2002). “[T]he optimum concentration [of ImpA] was about 15 mM where 6 monomer units were added to the primer in 24 hr. Use of 0.17 and 1.7 mM resulted in the addition of one and four monomer units, respectively. A 170 mM concentration of ImpA resulted mainly in the formation of the capped primer, Ap32pdA(pdA)8pA, and a small amount of 32pdA(pdA)8pApA.” Too much or too little of the montmorillonite was not good for the formation of long oligos either: 2 mg of clay in a 40 ml reaction mixture was just right; doubling that amount of clay slowed down the reaction; 10 mg of clay in 25 ml of reaction solution resulted in more efficient binding of the primer to the clay surface but less elongation of the primer.
It is possible that the seemly overperformance of MeadpA over ImpA is because Huang and Ferris used a very successful trick—to enrich and analyze only the long oligos (all short ones were removed)—because the vast majority of the products in reactions with MeadpA were dimers, trimers, and hydrolyzed monomers, just like reactions with ImpA (Ferris and Ertem 1993; Huang and Ferris 2003, 2006; Prabahar and Ferris 1997) (fig. 12).
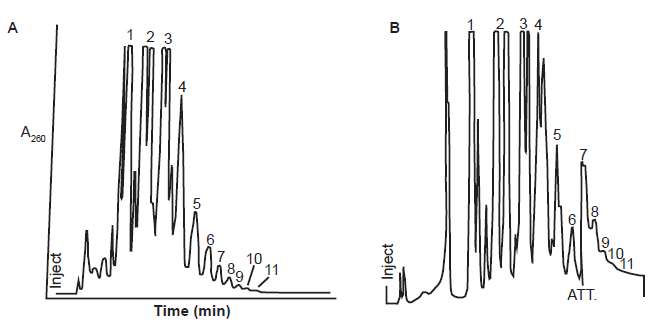
Fig. 12. Reaction products of MeadpA and ImpA on Na+ montmorillonite. (A) Anion exchange HPLC of the reaction products of MeadpA on Na+ montmorillonite. Fraction numbers in the figure correlate with the length of the linear oligomers contained in the corresponding fraction. (B) Anion-exchange HPLC of the reaction of ImpA on the Wyoming montmorillonite Na+ Volclay. Traced from Figure 1 of (Prabahar and Ferris 1997) (A) and Figure 1 of (Ferris and Ertem 1993) (B).
Note that the numbers in the high-performance liquid chromatography (HPLC, formerly referred to as high-pressure liquid chromatography) are fractions separated by the HPLC columns. It is common that each peak (or fraction) is a mixture of multiple chemicals because they cannot be separated in the experimental conditions (table 1). The desired products, if they can be generated in the reaction conditions, are just a minor product of the mixture. Note also that the yield of the polymers diminishes exponentially with the increase of number of its compositing monomers (fig. 12).
| Reaction Products | no NA+-Vol | with Na+-Vol | percentage | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ImpU | ImpI | ImpU | ImpI | ImpAa | ImpC | ImpG | MeadpA | |||
| The monomer fraction | 30 | 13 | 28/33 | 25 | 32 | percent of total | ||||
| pX | 90.5 | 89 | 90 | 89 | 100b | percent of the monomer fraction | ||||
| X5’ppX | 0.8 | 2.7 | 10 | 11 | 2.4c | |||||
| cyclic-X2’pX2’p | 76c | |||||||||
| cyclic-X2’pX3’p | 12c | |||||||||
| cyclic-X3’pX3’p | 8.5c | |||||||||
| The dimer fraction | 42 | 59 | 29/24 | 55 | 30 | percent of total | ||||
| pX2’pX | 1.6d | 2 | 28 | 2 | /33 | 73b | 20 | percent of the dimer fraction | ||
| pX3’pX | 0.7 | 6.4 | 9.5 | /52 | 26b | 45 | ||||
| X5’ppX2’pX | 2.2 | 2.4 | 4.4 | 0 | ||||||
| X5’ppX3’pX | 1.5 | 0 | /2.6 | 3 | ||||||
| cyclic-X3’pX3’pX3’p | 18 | 74 | /13e | 32 | ||||||
| cyclic-X3’pX2’pX3’p | 42 | 14 | ||||||||
| The trimer fraction | 0.2 | 0.1 | 12 | 12 | 18/18 | 12 | 17 | percent of total | ||
| pX2’pX2’pX | 30b | percent of the trimer fraction | ||||||||
| pX2’pX3’pX | 36b | |||||||||
| pX3’pX2’pX | /47 | 22b | 50 | |||||||
| pX3’pX3’pX | /28 | 18b | 37 | |||||||
| X5’ppX3’pX2’pX | /4 | 0 | ||||||||
| X5’ppX3’pX3’pX | /15 | 5 | ||||||||
| (Xp)mX5’ppX(pX)n’ (m+n=5) | /2 | 3 | ||||||||
| cyclic-X3’pX3’pX3’pX3’p | 0 | 5 | ||||||||
| tetramers | 6.2 | 7.9 | 13/11 | 6.1 | 11 | percent of total | ||||
| 5mers | 3.3 | 4.3 | 5.2/4.5 | 2.1 | 4 | |||||
| 6mers | 1.8 | 2.1 | 3.7/2.3 | 0.51 | 2.2 | |||||
| 7mers | 0.96 | 1.2 | 2.1/1.0 | 1.1 | ||||||
| 8mers | 1.1/.6 | 0.6 | ||||||||
| 9mers | 0.83/0.3 | 0.3 | ||||||||
| 10mers | 0.37/0.1 | 0.13 | ||||||||
| 11mers | /0.03 | 0.03 | ||||||||
Not all ImpN nucleotides react equally in the montmorillonite catalyzed reaction (Coari et al. 2017; Ertem and Ferris 2000; Miyakawa and Ferris 2003). “The rate of elongation of an RNA oligomer is determined by the 3’-nucleotide and the regiochemistry of its phosphodiester bond. The reaction rates are faster if there is a 3’-purine nucleotide and if this nucleotide is linked by a 3’,5’-phosphodiester bond. In contrast, an activated pyrimidine nucleotide adds more rapidly than the corresponding purine nucleotide to the 3’-end of the RNA oligomer” ((Miyakawa and Ferris 2003, 8206).
The relative rates of the initiation of dimer formation decrease in the order A > G > C > U by 8.4:4.8:1.3:1, respectively (Ertem and Ferris 2000). Furthermore, purine nucleotides, including ImpA, ImpI (5’-phosphorimidazolide inosine), and ImpG favor formation of 3’,5’ phosphodiester bonds, while pyrimidine nucleotides, such as ImpU and ImpC, favor formation of 2’,5’ phosphodiester bonds (Ferris and Ertem 1993; Kawamura and Ferris 1999; Miyakawa and Ferris 2003). The best reagent in this system is ImpI in that it has high regional selectivity and less side reactions (Kawamura and Ferris 1999). However, inosine is not a base used in cellular DNA or RNA.1
Therefore, clay-catalyzed polymerization of phosphoimidazolide-activated nucleotides does not constitute a condition to synthesize polynucleotides that are equivalent to natural DNA or RNA found in cells. It cannot generate long polynucleotides (The smallest free-living cell Mycoplasma genitalium has a genomic DNA of half a million base-pairs [Fraser et al. 1995]). In addition, the reaction system generates both 2’,5’ and 3’,5’ phosphodiester bonds in between nucleotides, often more 2’,5’ than 3’,5’ linkages. Furthermore, the incorporation of nucleotides is chemically-biased, resulting in oligomers with mostly purine bases, such as adenine, guanine, and inosine. Consequently, certain polynucleotide sequences may never be produced even if conditions forming long polynucleotides can be generated, greatly limiting diversity and information coding potential of resulted DNA or RNAs. Finally, unnatural nitrogenous bases, including inosine, can be easily incorporated in the polymers.
Using eutectic solutions
Another way to facilitate oligomerization of activated nucleotides is to have the reaction occur in eutectic solution, a solution with both water and ice (Monnard, Kanavarioti, and Deamer 2003; Kanavarioti, Monnard, and Deamer 2001). Oligos up to 17-mers can be synthesized from ImpN in such a condition. Like the reactions with metal ions or montmorillonite clay, the products are mainly hydrolyzed monomers, dimers, trimers, and tetramers, with traces of longer products (Ertem and Ferris 1996; Ferris and Ertem 1993; Kawamura and Ferris 1999; Monnard, Kanavarioti, and Deamer 2003).
The eutectic phase reaction is less purine-base-biased than the montmorillonite catalyzed reaction, with reactivity sequence A > G≈U > C (Monnard, Kanavarioti, and Deamer 2003). For an initial U:A:C:G 1:1:1:1 monomer mixture, the incorporation ratio is on average 1.24:1.61:1.00:1.28, or 1.29 purines for each pyrimidine. However, the formation of 2’,5’ phosphodiester bonds is favored over the formation of 3’,5’ phosphodiester bonds in eutectic solutions; although, the exact ratio has not been reported.
The concentration of the reactants is very important for the oligomerization of activated nucleotides in a eutectic solution; departure from the optimal ImpN concentrations by ±0.5 mM can result in lowering the reaction rates by up to a factor of two (Monnard and Szostak 2008).
Like the clay-catalyzed polymerization of phosphoimidazolide-activated nucleotides, the eutectic solution-mediated oligomerization does not constitute a condition to synthesize polynucleotides that are equivalent to natural DNA or RNA found in cells. The length of oligos formed in both conditions is extremely limited. The eutectic solution-mediated oligomerization is not as biased as the metal ion or clay-catalyzed oligomerization in terms of differential incorporation of different nucleotides, but oligomers formed in the eutectic reactions are more biased toward the formation of 2’,5’ phosphodiester bonds.
Using 3’, 5’ cyclic nucleotides as starting materials
A more recent development is to synthesize oligos using 3’,5’ cyclic nucleotides under dehydrating conditions, that is, using essentially dry materials (Costanzo et al. 2009, 2012, 2016; Morasch et al. 2014 Šponer et al. 2015). This oligomerization reaction is very efficient in generating oligos of GMP and AMP (oligoG and oligoA) but very inefficient in generating oligos of CMP and UMP (oligoC and oligoU).
This oligomerization reaction also requires a Goldilocks reaction condition. 3’, 5’ cyclic GMP oligomerizes efficiently to form oligoG of around 25-mers in length, but its concentration cannot be too low (<1 mM) or too high (>20 mM). The reaction temperature must be at or above 75oC in water (50oC under dehydrating conditions) but not too high. No sodium ion should be present (Costanzo et al. 2009, 2012; Šponer et al. 2015).
The yield of the reaction is pretty low and varies extensively depending on the conditions: 0.13% in a 85°C, 18 hours’ reaction, with 3’,5’ cyclic GMP concentration at 15mM, in water (Šponer et al. 2015). Under the same conditions, 3’,5’ cyclic AMP reacts hundreds of times slower, forming oligos of 4 to 8 nucleotides long (Costanzo et al. 2009). Afterward, these oligos and their ligation products ligate to form heterogeneous polyA up to 120 nucleotides long. However, 3’,5’ cyclic CMP and 3’,5’ cyclic UMP forms oligos with an average length of 5.5 nucleotides.
Later reports from the same laboratory state that “Oligomerization of 3’, 5’ cGMP produces up to 20-mers, whereas the longest oligomer obtained starting from 3’,5’ cAMP was a 4-mer” (Costanzo et al. 2016), and that “heat-induced oligomerizations conducted under the same conditions did not lead to detectable amount of oligomers when starting from dry samples of 3’,5’-cCMP” (Costanzo et al. 2017, 1536). To make oligoC, Costanzo and colleagues used a temperature of 20oC and proton or UV irradiation, and the oligos formed were only 2 to 3 nucleotides long (Costanzo et al. 2017). The discrepancies are probably caused by the sensitivity of the oligomerization on reaction conditions, including concentrations of monomers, temperatures, pH, reaction time, and trace amounts of sodium ions.
It is remarkable that, as ardent advocates of “Molecular Darwinism” and the one-pot proton-irradiated reaction to generate all the basic building blocks of life, Di Mouro and his colleagues never start their oligomerization reactions with the products generated in their one-pot reactions but with highly purified 3’,5’ cyclic NMPs (N: A, G, C, or U) that are often “custom-made and specially purified in order to guarantee (i) the maximal possible purity relative to the absence of adducts-forming cations (mostly Na+) and (ii) the absence of evaporation or precipitation steps during the course of the whole process” (Costanzo et al., 2012, 2016, 2017; Morasch et al. 2014; Saladino et al. 2018; Šponer et al. 2015). They did this because they were fully aware of the problem of Benner’s “Asphalt Paradox” (Benner 2014). In Di Mouro et als’ own words: “As Benner noted, prebiotic chemistry without selection leads to tar formation. As several literature examples on the oligomerization of HCN demonstrate, this statement is especially true for radical chemistry” (Saladino et al. 2018).
Therefore, neither the use of various inorganic catalysts nor eutectic phase nor 3’,5’ cyclic nucleotides can provide a condition to synthesize polynucleotides that are equivalent to cellular DNA or RNA. None of them can generate polynucleotides the length of cellular DNA or RNA. With the first two methods mentioned above, using ImpN or similarly activated nucleotides as starting materials, both 2’,5’ and 3’,5’ phosphodiester bonds form in between nucleotides, often mainly 2’,5’ phosphodiester bonds. Furthermore, the sequences of the polynucleotides are restricted by the characteristics of the nucleotides, with a bias toward incorporating purine rather than pyrimidine nucleotides, and the pre-activated nucleotides have to be synthesized in organic solvents, in the absence of water (Template-directed incorporation of nucleotides upon in situ activation has been described by (Jauker, Griesser, and Richert 2015). OligoA of the length of 9-mers can be formed in such conditions). The third method uses 3’, 5’ cyclic nucleotides that are more stable and are easier to synthesize than ImpN, but incorporation of uridine and cytosine in the polymerization reaction is problematic. A more serious problem is the absence of plausible prebiotic synthesis and maintenance of the requisite activated, unstable, mononucleotides.
The problem of homochirality
Not surprisingly, activated D-ribose nucleotides and L-ribose nucleotides behave equally in all the non-templated oligonucleotide synthesis reactions tested (Joyce et al. 1984; Osawa, Urata, and Sawai 2005; Urata et al. 2001). The tendency of a nucleotide joining with another nucleotide that has the same chirality to form homochiral oligomers or joining with one that has the opposite chirality depends on the characteristics of the nucleotides and their concentrations as well as the presence of other chemicals (Joshi, Pitsch, and Ferris 2000, 2007; Osawa, Urata, and Sawai 2005; Urata et al. 2001). Thus, at a concentration of 0.60 mM (2hr reaction), D,L-ImpA forms dimers of which 94% are D,D and L,L and 6% D,L and L,D (Joshi, Pitsch, and Ferris 2000).
Increasing the concentration of D,L-ImpA to 4.8 mM (35 days reaction), the percentage of D,D and L,L dimers decreases to 50.8% (Joshi, Pitsch, and Ferris 2007). The addition of montmorillonite has a similar effect as increasing the concentration of D,L-ImpA. In the presence of montmorillonite, D,L-ImpA (0.60 mM, 2hr reaction) forms dimers with about 60% of D,D and L,L dimers (Joshi, Pitsch, and Ferris 2000, 2007). ImpU (0.60 mM, 1.5hr reaction) forms mostly (59.4%) heterodimers (Joshi, Pitsch, and Ferris 2007). In the absence of montmorillonite, ImpU (4.8 mM, 30 days reaction) forms mostly (84.2%) D,D and L,L homodimers. The percentages of homotrimers are much lower than those of homodimers for both ImpA and ImpU. In the presence of lead ion or uranyl ion, 50mM of racemic D,L-ImpA dimerize to form mainly homodimers, 54% and 65%, respectively (Osawa, Urata, and Sawai 2005).
The higher percentage of homodimers (DD and LL) compared to heterodimers (DL and LD) in some of the reaction conditions is mistakenly praised as a way of solving the famous homochiral problem because even the homodimers are, in fact, a racemic mixture, with inseparable DD and LL, while homochiral in cells refers to purely DDs in DNA and RNA.
Templated non-enzymatic oligonucleotide formation
Would the presence of DNA or RNA templates resolve the issues of oligos generated in non-enzymatic reactions being too short, linked with the wrong phosphodiester bonds, and with chemically-biased base composition?
The short answer is: NO.
The addition of premade polynucleotide templates that consist homochiral nucleotides, linked with 3’,5’ phosphodiester bonds, facilitates the non-enzymatic oligomerization of the activated nucleotides that have the same chirality as the construction units of the templates, forming short oligos with both 3’,5’ and 2’,5’ phosphodiester linkages (Sulston et al. 1968; Weimann et al. 1968). The polynucleotide templates offer little help for the oligomerization of the activated nucleotides that have opposite chirality.
For example, with polyU as a template, AMP oligomerizes in the presence of the dehydrating agent CDI (1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride) (Sulston et al. 1968) (table 2 and fig. 13). The desired pA3’p5’A was produced with a yield of 2.5%. Other reaction products included pA2’p5’A (14%), A5’pp5’A (25%), and A5’pp5’ApA (6%).
| Starting nucleotide* | Template | Polymers | Dimers (yield) | ||||
|---|---|---|---|---|---|---|---|
| Length | Ratio of 3’-5’ diester bond | 3’-5’ diester bond | 2’-5’ diester bond | 5’-5’ diester bond | 5’pp5’ pyrophosphate | ||
| pA | poly U | one or more trimersb | pA3’p5’A (2.5%) | pA2’p5’A (14%) | A5’pp5’A (25%) A5’pp5’ApA (6%) |
||
| no | less than 1% of (pApA + A5’pp5’ApA) | A5’pp5’A (25%) | |||||
| pA and A | poly U | trimers ApApA (1.4%) | 18% | A3’p5’A (0.3%) | A2’p5’A (10%)td> | A5’p5’A (5.4%) | |
| no | Without poly U, reaction much less efficient | ||||||
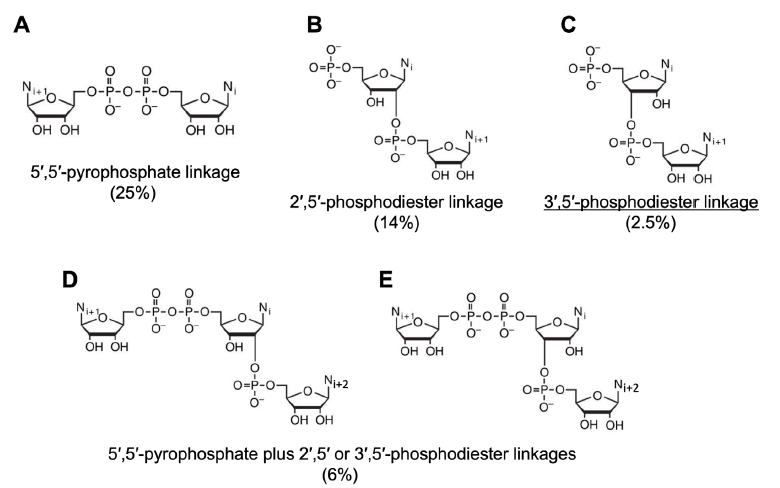
Fig. 13. Some products of polyU-templated AMP polymerization and their yields. The chemical structures are derived from Figure 1A of (Robertson and Joyce 2012). Yield percentages are from (Sulston et al. 1968). The product with the correct linkage is underlined.
Note that pA3’p5’A, the molecule with the correct linkage, that is, nucleotides linked by 3’,5’ phosphodiester bonds, has the lowest yield in the reported four products of CDI-facilitated AMP condensing reaction. Without poly U, the same reaction produced about 25% of A5’pp5’A and less than 1% of (pApA + A5’pp5’ApA).
Addition of adenosine, which is produced when AMP hydrolyzes, in the starting materials reduced the yield of pA3’p5’A. ApApA trimer was formed at a yield of 1.4% in this condition. Eighteen percent of the linkages inside the trimers are 3’,5’ phosphodiester bonds. As when AMP was used as the starting material, the reaction was much less efficient without templates.
It is striking that even though the starting templates have homolinkages—all nucleotides are linked via 3’,5’ phosphodiester bonds—the products have a mixture of linkages, of which most are not the desired 3’,5’ phosphodiester bonds (Sulston et al. 1968) (table 2, fig. 13).
The ratio of 3’,5’- to 2’,5’-phosphodiester linkages of the products varies depending on the characteristics of the templates, the nitrogenous bases of the nucleotides, and the activation groups of the nucleotides (Ertem and Ferris 1996; Grzeskowiak and Orgel 1986; Inoue and Orgel 1983; Joyce 1984; Joyce, Inoue, and Orgel 1984; Joyce and Orgel 1988; Prakash, Roberts, and Switzer 1997; Sulston et al. 1968). Unlike oligomerization using polyU as templates, oligoG formed using polyC templates contains mainly 3’,5’ phosphodiester bonds (Ertem and Ferris 1996; Grzeskowiak and Orgel 1986).
There are several problems for templated non-enzymatic oligomerizations:
First, to state the obvious, templated oligonucleotide formation begs the question of where the first template comes from.
Second, unlike protein enzymes-dependent DNA or RNA formation, not all oligos can serve as templates for non-enzymatic oligomerization (Hey, Hartel, and Gobel 2003; Inoue and Orgel 1983; Joyce 1984; Joyce, Inoue, and Orgel 1984; Joyce and Orgel 1988; Kozlov et al. 1999; Orgel 2004; Stribling and Miller 1991; Wu and Orgel 1992a, 1992b, 1992c). C- and G-rich templates allow effective primer elongations with high fidelity, while polyA templates do not. The best templates are C-rich oligos, runs of G residues might be present, but A and T residues have to be isolated from each other and from G residues. The sequences TT, GT, and TG interfere with copying partially. The sequences AT, TA, AA, GA, and AG are almost total barriers.
Consequently, Wu and Orgel commented: “A minimal self-replicating system consists of two complementary sequences each of which is capable of facilitating the synthesis of the other. Is such a system possible within the framework of our model system? Clearly a copolymer of C and G cannot qualify. If the two complements contain roughly equal amounts of C and G they are disqualified because they give rise to extensive intramolecular self-structure. If one polymer is rich in C, the other must be rich in G and so is disqualified because it forms a stable intermolecular tetrahelical self-structure. Can the introduction of isolated A and T residues save the situation? This seems unlikely, but one cannot be sure without more experimental information on the efficiency of template directed synthesis on oligoG sequences that contain an isolated A or T residue, for example, -GGAGG- and related sequences” (Wu and Orgel 1992b).
Twenty years later, Loffler and colleagues had some, but very limited, success in extension primers across the AA, AU, and AG blockers (Loffler et al. 2013) by incubating the reaction mixtures below their freezing temperature, but above their eutectic point (that is, in eutectic solutions); the template-directed primer extension had low yield and was prone to nucleotide misincorporation.
Third, the presence of 2’,5’ phosphodiester bonds in a polynucleotide made of mainly 3’,5’-phosphodiester bonds reduces its ability to act as a template, destabilizes the templates, and increases 2’,5’ phosphodiester bonding between nucleotides (Ertem and Ferris 1996; Lohrmann and Orgel 1978; Prakash, Roberts, and Switzer 1997; Sawai 1988; Sinha, Kim, and Switzer 2004; Usher and McHale 1976). However, as discussed above, 2’,5’ phosphodiester linkage is often the predominant product of untemplated, nonenzymatic, oligomerizations of nucleotide monomers.
Fourth, the homolinkage of nucleotides in the starting materials cannot be propagated; polymerization using templates that are made of homochiral nucleotides and 3’,5’ phosphodiester linkages produces oligomers that contains not only 3’,5’ phosphodiester linkages but also 2’,5’ phosphodiester linkages.
Fifth, the presence of the opposite chiral isomers shuts down template-directed formation of oligoribonucleotides, at least for both polyU and polyC templates (Joyce, Inoue, and Orgel 1984; Kozlov, Pitsch, and Orgel 1998; Osawa, Urata, and Sawai 2005); although, the presence of the opposite chiral isomers of ribonucleotides only slightly inhibits the un-templated formation of oligoribonucleotides; e.g., changing the length of the longest oligomers from 8 to 7 (Joshi, Pitsch, and Ferris 2000, 2007; Osawa, Urata, and Sawai 2005; Urata et al. 2001).
Therefore, even if a homochiral RNA or DNA polymer were synthesized in a prebiotic condition, its replication would be problematic. The presence of enantiomers would shut down the replication of any homochiral polynucleotides; yet, the presence of enantiomers is unavoidable because it is chemically and physically impossible to generate homochiral monomers in an achiral environment (Joyce, Inoue, and Orgel 1984). Besides, replication of an efficient template, such as polyC, generates a complementary strand—polyG—that is an inefficient template (Hill, Orgel, and Wu 1993). In addition, replication of a polynucleotide with purely 3’,5’ phosphodiester bonds produces a complementary polynucleotide containing 2’,5’ phosphodiester bonds, which reduce the templating ability of the resulted polynucleotide. Lastly, all templated, non-enzymatic, oligonucleotide formation reactions have low yields, and polynucleotides containing adenine and uracil cannot be fully copied.
Helping with short RNA helpers?
Two reasons for the lack of a feasible method to non-enzymatically copy sequences containing all four natural nucleotides are the slow rate of primer extension with adenosine and uridine monomers and the inhibition of the oligomerization of the activated nucleotides by the free un-activated nucleotides, which are the hydrolyzed products of the activated starting nucleotides (Deck, Jauker, and Richert 2011; Joyce et al. 1987; Sosson and Richert 2018; Wu and Orgel 1992b).
By immobilizing templates and primers on magnetic iron oxide beads and periodically replacing reaction solutions, and using short (3–4 nucleotides long) downstream-binding micro-helpers, RNAs whose sequences are complementary to the template, all four nucleobases (A, U, G, C) in the templates were copied efficiently at 0°C (Deck, Jauker, and Richert 2011; Sosson and Richert 2018; Vogel and Richert 2007) (fig. 14).
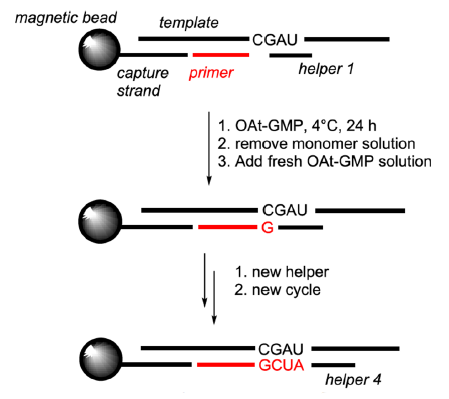
Fig. 14. RNA copying in the presence of micro-helpers. From Figure 10 of (Sosson and Richert 2018). OAtdNMPs (OAt-GMP, specifically, in the image) represent oxyazabenzotriazolides of deoxynucleotides. Openaccess article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0).
The primer extension rate was greatly increased by using activated helper oligos (Prywes et al. 2016). In addition, with the aid of the helpers, the reaction rate differences between primer extension with G or C monomers and that with A and U decreased from three orders of magnitude to about one. Replacing the canonical U monomer with 2-thiouridine and 2-thoribothymine made the primer extension rate comparable to that with G or C addition in such reactions.
There are two caveats for this kind of reaction: (1) the instantaneous replacement of the spent monomers with activated ones, and (2) the instantaneous, sequential, displacement of used helpers with new ones with the correct sequences as demanded by the sequence of the template. Indeed, overlapping helping trimers competed with each other for binding sites on the template and slowed down the total primer extension rate and efficiency, so that only sequences around seven basepairs long could be copied in one reaction in low yields (Prywes et al. 2016). Only by the sequential addition of the activated monomers, along with their attendant activated trimers, one at a time, could longer RNA fragments be synthesized.
Starting with trinucleotides?
Attwater and colleagues reported a very creative RNA copying system that uses activated trinucleotides, instead of mononucleotides, as basic building blocks (Attwater et al. 2018). This system is powered with a ribozyme (RNA catalyst) made of a 153 nucleotides long catalytic RNA and a 135 nucleotides long helper RNA. The system allows copying of RNA templates containing all four nucleobases and even copying of RNA templates with secondary structures.
The abiotic relevance of this system is questionable because the unlikely abiotic origins of the templates, of the activated trinucleotides, and, especially, of the ribozyme. As shown in the abovementioned experiments, it is unlikely for an abiogenic process to generate the long and complicated ribozyme subunits that has been artificially synthesized and artificially selected by the authors. Furthermore, in an abiotic world, the only way of generating the required ribozyme would be through self-linking of nucleotides, or similar chemicals. However, self-linking molecules destroy genetic information coding and decoding systems, including DNA (or RNA) replication, transcription, and translation systems, as discussed in Tan (2022a).
Lessons from top-down studies
In living cells, all nucleotides in DNA and RNA are linked via 3’,5’ phosphodiester bonds. Each base can be followed or preceded by any other base regardless of its chemical property, just like the bits and bytes of information on a computer hard disk are free to form any sequence without physical or chemical bias. This characteristic of DNA (and computer hard disks) is critical, in fact essential, for DNA to serve as the genetic information carrier. A DNA strand normally forms an antiparallel double-stranded helix with another DNA strand that has a reverse and complementary sequence, in that A, T, G, C of one strand form hydrogen bonds with T, A, C, G of its complementary strand. In addition, whether the double-stranded DNA in a cell is replicated, and if so, when and how it is replicated is determined by the proteins and RNAs inside the cell and the status of the cell and its environment. Both under- and over-replication cause genome instability.
When a DNA molecule is replicated, the replication machinery will make a copy with almost the exact sequence as the parental DNA. Each parental strand serves as a template to make a reverse and complementary daughter strand. Therefore, as long as one knows the sequence of one strand, the sequence of the other can be spelled out according to the A-T, G-C base pairing rule.
This gives rise to the popular and long-standing misconception that DNA self-replicates and that the A-T and G-C base pairing force determines the accuracy of copying.
The truth is that proteins replicate the DNA, and the accuracy of replication is determined by the fidelity of the DNA polymerase, the concentration and ratio of nucleotides, and the concentration of Mg2+. Different DNA polymerases have different fidelity. For example, polymerase Pfu, a DNA polymerase isolated from the hyperthermophilic archaeon Pyrococcus furiosus, is much more accurate than polymerase Taq, a DNA polymerase isolated from the thermophilic bacterium Thermus aquaticus. Furthermore, the error rates of a polymerase can be increased by reducing the percentage of one of the four deoxyribonucleotides needed to synthesize a DNA molecule, by increasing the concentration of Mg2+, or by substituting Mg2+ with Mn2+ (manganese) during PCR (polymerase chain reaction). The error rate can also be increased when dnaQ, the ε subunit of E. coli DNA polymerase III, is mutated. Finally, a polymerization reaction can be halted completely when some protein components of the replication machinery stop functioning. For example, E. coli DNA polymerization stops almost immediately when several E. coli genes are mutated, such as dnaB (a helicase), dnaE (α subunit of DNA polymerase III), ssb (single-stranded DNA binding protein), gyrA and gryB (subunits of DNA gyrase) (Tropp 2012, 380). Therefore, the sequence of the generated DNA is not ultimately determined by the chemical or physical affinities of nucleotides, nor the A-T, G-C base-pairing rules.
It is ironic that even though a multitude of proteins are required for DNA replication, these proteins do not ultimately determine the sequence of the DNA they make, either. Even the cells hosting both the DNA and proteins do not have the final word. Otherwise, no molecular cloning would be possible, and no GMO (genetically modified organisms) would have been generated.
That said, cells that have a functional replication machinery can replicate DNA molecules if these molecules contain a replication origin that can be recognized as such by the replication machinery, making daughter DNA with a sequence that matches the sequence of the parental DNA. Where did the first parental DNA come from? That is the question.
Note that the problems of water, asphalt, homochirality, and homolinkage are not at all problems for cells, but for abiogenesis, that is, synthesizing biomolecules outside of naturally existing organisms (Tan and Stadler 2020). Furthermore, an astounding discovery of molecular biology is the organism-specific genetic information coding and decoding systems, which this author addressed a few years ago (Tan and Tomkins 2015a, 2015b) and will be further explored in Tan 2022c.
Conclusion
Theoretical considerations and both the bottom-up and the top-down experimental studies suggest that, for the first cell, chemistry alone is incapable of creating the first strand of nucleic acid, be it a DNA or an RNA molecule, for the first cell.
References
Attwater, James, Aditya Raguram, Alexy S. Morgunov, Edoardo Gianni, and Philipp Holliger. 2018. “Ribozyme-Catalysed RNA Synthesis Using Triplet Building Blocks.” Elife 7. https://elifesciences.org/articles/35255.
Bardi, Jason Socrates. 2004. “Study Suggests Component of Volcanic Gas May Have Played a Significant Role in the Origins of Life on Earth.” News and Views 3, no. 30. https://www.scripps.edu/newsandviews/e20041011/ghadiri.html.
Benner, Steven A. 2014. “Paradoxes in the Origin of Life.” Origins of Life and Evolution of Biospheres 44, no. 4 (22 January): 339–343.
Coari, Kristin M., Rebecca C. Martin, Kopal Jain, and Linda B. McGown. 2017. “Nucleotide Selectivity in Abiotic RNA Polymerization Reactions.” Origins of Life and Evolution of Biospheres 47, no. 3 (3 February): 305–321.
Costanzo, Giovanna, Samanta Pino, Fabiana Ciciriello, and Ernesto Di Mauro. 2009. “Generation of Long RNA Chains in Water.” Journal of Biological Chemistry 284, no. 48 (November): 33206–33216.
Costanzo, Giovanna, Raffaele Saladino, Giorgia Botta, Alessandra Giorgi, Anita Scipioni, Samanta Pino, and Ernesto Di Mauro. 2012. “Generation of RNA Molecules by a Base-Catalysed Click-Like Reaction.” ChemBioChem 13, no. 7 (7 May): 999–1008.
Costanzo, Giovanna, Samanta Pino, Anna Maria Timperio, Judit Šponer, Jiří Šponer, Olga Nováková, Ondrej Šedo, Zbyněk Zdráhal, and Ernesto Di Mauro. 2016. “Non-Enzymatic Oligomerization of 3’, 5’ Cyclic AMP.” PLoS One 11, no. 11 (1 November): e0165723. https://doi.org/10.1371/journal.pone.0165723.
Costanzo, Giovanna, Alessandra Giorgi, Anita Scipioni, Anna Maria Timperio, Carmine Mancone, Marco Tripodi, Michail Kapralov, et al. 2017. “Nonenzymatic Oligomerization of 3’,5’-Cyclic CMP Induced by Proton and UV Irradiation Hints at a Nonfastidious Origin of RNA.” Chembiochem 18, no. 15 4 August)): 1535–1543.
Deck, Christopher, Mario Jauker, and Clemens Richert. 2011. “Efficient Enzyme-Free Copying of all Four Nucleobases Templated by Immobilized RNA.” Nature Chemistry 3, no. 8 (10 July): 603–608.
Ding, Ping. Z., Kunio Kawamura, and James P. Ferris. 1996. “Oligomerization of Uridine Phosphorimidazolides on Montmorillonite: A Model for the Prebiotic Synthesis of RNA on Minerals.” Origins of Life and Evolution of the Biosphere 26, no. 2 (April): 151–171.
Ertem, Gözen, and James P. Ferris. 1996. “Synthesis of RNA Oligomers on Heterogeneous Templates.” Nature 379 , no. 6562 (18 January): 238–240.
Ertem, Gözen, and James P. Ferris. 1997. “Template-Directed Synthesis Using the Heterogeneous Templates Produced by Montmorillonite Catalysis. A Possible Bridge Between the Prebiotic and RNA Worlds.” Journal of the American Chemical Society 119, no. 31 (6 August)): 7197–7201.
Ertem, Gözen, and James P. Ferris. 2000. “Sequence- and Regio-Selectivity in the Montmorillonite-Catalyzed Synthesis of RNA.” Origins of Life and Evolution of the Biosphere 30, no. 5 (October): 411–422.
Ferris, James P. 2002. “Montmorillonite Catalysis of 30–50 mer Oligonucleotides: Laboratory Demonstration of Potential Steps in the Origin of the RNA World.” Origins of Life and Evolution of the Biosphere 32, no. 4 (August): 311–332.
Ferris, James P., and Gözen Ertem. 1992. “Oligomerization of Ribonucleotides on Montmorillonite: Reaction of the 5’-Phosphorimidazolide of Adenosine.” Science 257, no. 5075 (4 September): 1387–1389.
Ferris, James P., and Gözen Ertem. 1993. “Montmorillonite Catalysis of RNA Oligomer Formation in Aqueous Solution. A Model for the Prebiotic Formation of RNA.” Journal of the American Chemical Society 115, no. 26 (1 December): 12270–12275.
Ferris, James P., Aubrey R. Hill, Jr., Rihe Liu, and Leslie E. Orgel. 1996. “Synthesis of Long Prebiotic Oligomers on Mineral Surfaces.” Nature 381, no. 6577 (2 May): 59–61.
Fialho, David M., Suneesh C. Karunakaran, Katherine W. Greeson, Isaac Martinez, Gary B. Schuster, Ramanarayanan Krishnamurthy, and Nicholas V. Hud. 2021. “Depsipeptide Nucleic Acids: Prebiotic Formation, Oligomerization, and Self-Assembly of a New Proto-Nucleic Acid Candidate.” Journal of the American Chemical Society 143, no. 34 (1 September): 13525–13537.
Fraser, Claire M., Jeannine D. Gocayne, Owen White, Mark D. Adams, Rebecca A. Clayton, Robert D. Fleischmann, Carol J. Bult et al. 1995. “The Minimal Gene Complement of Mycoplasma genitalium.” Science 270, no. 5235 (20 October): 397–403.
Grzeskowiak, Kazimierz, and Leslie E. Orgel. 1986. “Template-Directed Synthesis on Short Oligoribocytidylates.” Journal of Molecular Evolution 23, no. 4 (December): 287–279.
Hey, Marcus, Christian Hartel, and Michael W. Göbel. 2003. “Nonenzymatic Oligomerization of Ribonucleotides: Towards in vitro Selection Experiments.” Helvetica Chimica Acta 86, no. 3 (March): 844–854.
Hill, Aubrey R. Jr., Leslie E. Orgel, and Taifeng Wu. 1993. “The Limits of Template-Directed Synthesis with Nucleoside-5’-Phosphoro(2-Methyl)Imidazolides.” Origins of Life and Evolution of the Biosphere 23, nos. 5–6 (December): 285–290.
Huang, Wenhua, and James P. Ferris. 2003. “Synthesis of 35–40 Mers of RNA Oligomers from Unblocked Monomers. A Simple Approach to the RNA World.” Chemical Communications (Cambridge, England) 12 (21 June): 1458–1459.
Huang, Wenhua, and James P. Ferris. 2006. “One-Step, Regioselective Synthesis of up to 50-Mers of RNA Oligomers by Montmorillonite Catalysis.” Journal of the American Chemical Society 128, no. 27 (1 July): 8914–8919.
Inoue, T., and L. E. Orgel. 1983. “A Nonenzymatic RNA Polymerase Model.” Science 219, no. 4586 (18 February): 859–862.
Jauker, Mario, Helmut Griesser, and Clemens Richert. 2015. “Copying of RNA Sequences without Pre-Activation.” Angewandte Chemie-International Edition 54, no. 48 (23 November): 14559–14563.
Joshi, Prakash C., Stefan Pitsch, and James P. Ferris. 2000. “Homochiral Selection in the Montmorillonite-Catalyzed and Uncatalyzed Prebiotic Synthesis of RNA.” Chemical Communications, 24 (29 November): 2497–2498.
Sawai, H. 1976. “Catalysis of Internucleotide Bond Formation by Divalent Metal Ions.” Journal of the American Chemical Society 98, no. 22 (1 October): 7037–7039.
Sawai, H. 1988. “Oligonucleotide Formation Catalyzed by Divalent Metal Ions. The Uniqueness of the Ribosyl System.” Journal of Molecular Evolution 27, no. 3 (May): 181–186.
Sawai, Hiroaki, Katsutaka Higa, and Kensei Kuroda. 1992. “Synthesis of Cyclic and Acyclic Oligocytidylates by Uranyl-Ion Catalyst in Aqueous-Solution.” Journal of the Chemical Society, Perkin Transactions 1, no. 4: 505-508.
Sawai, Hiroaki, Kensei Kuroda, and Takashi Hojo. 1989. “Uranyl-Ion as a Highly Effective Catalyst for Internucleotide Bond Formation.” Bulletin of the Chemical Society of Japan 62, no. 6: 2018–2023.
Sinha, Surajit, Paul H. Kim, and Christopher Switzer. 2004. “2’,5’-linked DNA is a Template for Polymerase-Directed DNA Synthesis.” Journal of the American Chemical Society 126, no. 1 (February): 40–41.
Sosson, Marilyne, and Clemens Richert. 2018. “Enzyme-Free Genetic Copying of DNA and RNA Sequences.” Beilstein Journal of Organic Chemistry 14 (March 12): 603–617.
Šponer, Judit E., Jiří Šponer, Alessandra Giorgi, Ernesto Di Mauro, Samanta Pino, and Giovanna Costanzo. 2015. “Untemplated Nonenzymatic Polymerization of 3’, 5’cGMP: A Plausible Route to 3’,5’-Linked Oligonucleotides in Primordia.” Journal of Physical Chemistry B 119, no. 7 (27 January): 2979–2989.
Stribling, Roscoe, and Stanley L. Miller. 1991. “Attempted Nonenzymatic Template-Directed Oligomerizations on a Polyadenylic-Acid Template: Implications for the Nature of the First Genetic Material.” Journal of Molecular Evolution 32, no. 4 (April): 282–288.
Sulston, J., R. Lohrmann, L. E. Orgel, and H. Todd Miles. 1968. “Nonenzymatic Synthesis of Oligoadenylates on a Polyuridylic Acid Template.” Proceedings of the National Academy of Sciences USA 59, no. 3 (March): 726–733.
Tan, Change Laura, and Rob Stadler. 2020. The Stairway To Life: An Origin-Of-Life Reality Check. Evorevo Books.
Tan, Change, and Jeffrey P. Tomkins. 2015a. “Information Processing Differences Between Archaea and Eukaraya—Implications for Homologs and the Myth of Eukaryogenesis.” Answers Research Journal 8 (March 18): 121–141. https://answersingenesis.org/biology/microbiology/information-processing-differences-between-archaea-and-eukarya/.
Tan, Change, and Jeffrey P. Tomkins. 2015b. “Information Processing Differences Between Bacteria and Eukarya—Implications for the Myth of Eukaryogenesis.” Answers Research Journal 8 (March 25): 143–162. https://answersingenesis.org/biology/microbiology/information-processing-differences-between-bacteria-and-eukarya/.
Tan, Change Laura. 2022a. “Facts Cannot be Ignored When Considering the Origin of Life #1: The Necessity of Biomonomers Not to Self-Link for the Existence of Living Organisms.” Answers Research Journal 15 (9 March): 25–39. https://answersresearchjournal.org/genetics/necessity-of-bio-monomers/.
Tan, Change Laura. 2022c. “Facts Cannot be Ignored When Considering the Origin of Life #3: Necessity of Matching the Coding and the Decoding Systems.” Answers Research Journal 15 (in press).
Tropp, Burton E. 2012. Molecular Biology: Genes to Proteins. 4th ed. Sudbury, Massachusetts: Jones & Bartlett Learning.
Urata, Hidehito, Chie Aono, Norihiko Ohmoto, Yuko Shimamoto, Yoshiko Kobayashi, and Masao Akagi. 2001. “Efficient and Homochiral Selective Oligomerization of Racemic Ribonucleotides on Mineral Surface.” Chemistry Letters 30, no. 4 (April): 324–325.
Usher, D. A., and A. H. McHale. 1976. “Hydrolytic Stability of Helical RNA: A Selective Advantage for the Natural 3’,5’-Bond.” Proceedings of the National Academy of Sciences USA 73, no. 4 (April): 1149–1153.
Vogel, Stephanie R., and Clemens Richert. 2007. “Adenosine Residues in the Template Do Not Block Spontaneous Replication Steps of RNA.” Chemical Communications 19: 1896–1898.
Weimann, B. J., R. Lohrmann, L. E. Orgel, H. Schneider-Bernloehr, and J. E. Sulston. 1968. “Template-Directed Synthesis With Adenosine-5’-Phosphorimidazolide.” Science 161, no. 3839 (26 July): 387.
Wu, T., and L. E. Orgel. 1992a. “Nonenzymatic Template-Directed Synthesis on Hairpin Oligonucleotides. 2. Templates Containing Cytidine and Guanosine Residues.” Journal of the American Chemical Society 114, no. 14 (1 January): 5496–5501.
Wu, T., and L. E. Orgel. 1992b. “Nonenzymatic Template-Directed Synthesis on Hairpin Oligonucleotides. 3. Incorporation of Adenosine and Uridine Residues.” Journal of the American Chemical Society 114, no. 21 (7 October): 7963–7969.
Wu, T., and L. E. Orgel. 1992c. “Nonenzymatic Template-Directed Synthesis on Oligodeoxycytidylate Sequences in Hairpin Oligonucleotides.” Journal of the American Chemical Society 114, no. 1 (1 January): 317–322.