
The views expressed in this paper are those of the writer(s) and are not necessarily those of the ARJ Editor or Answers in Genesis.
Abstract
The current chimpanzee genome assembly has problems that reduce its veracity as an authentic representation. First, it has been assembled using the human genome as a reference scaffold and does not stand on its own merits. Second, given the fact that significant levels of human DNA exist in non-primate databases due to laboratory and worker contamination, the potential for human DNA in the pre-assembled chimpanzee sequencing reads is highly probable. Therefore, 101 Sanger-style publically available trace read data sets were downloaded, end-trimmed for low quality bases, and purged of vector sequence. Then, 25,000 sequences were selected at random from each of the 101 data sets and queried against the human genome using BLASTN v2.2.31 with gap extension. Results from the BLASTN analysis indicated that two different groups of chimpanzee DNA sequences could be found. Those that were completed early in the chimpanzee genome project that contributed to the initial 5-fold draft genome, were considerably more similar to human than those that were produced later in the project by a difference of about 7% overall data set identity and produced 6% less hits onto the human genome. Sequences (both alignable and non-alignable) from the seemingly less contaminated data sets indicate that the chimpanzee genome is approximately 85% identical overall to human. Extensive poor alignment of chimpanzee DNA sequences that did not have hits on the human genome that were blasted on the chimpanzee genome revealed regions of miss-assembly for the chimpanzee genome.
Introduction
One of the problems with the current status of the chimpanzee genome is that it has not been constructed on its own merits through the use of an accurate integrated physical-genetic map (Tomkins 2011). Instead, all of the short DNA sequences produced by the DNA sequencing machinery (known as trace reads) have been assembled onto the human genome—using it as a framework scaffold or reference sequence (Mikkelsen et al. 2005; Prado-Martinez et al. 2013; Tomkins 2011). This was done out of budget constraints, convenience, and a healthy dose of evolutionary presupposition that humans evolved from apes.
Another serious potential problem with the chimpanzee genome is the issue of human DNA contamination that would also result in the production of a more human-like chimpanzee genome. In 2011, a very interesting study was published in which the researchers screened 2749 non-primate public DNA databases and found 492 to be contaminated with human sequence at levels of up to 10% (Longo, O’Neill, and O’Neill 2011). The contaminated DNA databases represented species ranging from bacteria to plants to fish. Ape and monkey databases were not tested, leaving the question open as to how much human DNA contamination may be present in them. The sequencing of archaic human DNA such as Neandertal has also been plagued with the problem of modern human DNA contamination—leading to the recent development of strict laboratory precautions (Skoglund et al. 2014; Thomas and Tomkins 2014). Nevertheless, modern human DNA contamination is a standard problem in earlier published ancient DNA studies (Noonan 2010; Skoglund et al. 2014; Thomas and Tomkins 2014). In light of results from these studies combined with the fact that the DNA sequencing that led to the 2005 rough draft of the chimpanzee genome was produced during an era in which the problem of human DNA contamination was not yet adequately realized or appreciated, the potential for human DNA contamination in the chimpanzee genome is a valid possibility.
Given that both a biased sequence assembly using the human genome as a framework combined with the distinct possibility of human DNA contamination may very well have led to the development of a chimpanzee genome that is more human-like than it should be, research was initiated to assess characteristics of chimpanzee Sanger-style trace read DNA sequences produced between the years 2000 and 2011. One future goal of this effort would be to identify DNA sequence datasets having indicators of reduced levels of human DNA contamination for a reassembly of the chimpanzee genome without the use of the human genome as a reference. This is called a de novo assembly, meaning that no reference genome is used (Bradnam et al. 2013; Narzisi and Mishra 2011). The end result will be a genome that is more accurate, but considerably less contiguous than one assembled using a reference sequence (Henson, Tischler, and Ning 2012).
Materials and Methods
Chimpanzee Sanger-style trace read files, their corresponding quality files, and xml files containing sequencing run information were downloaded from the NCBI trace read archive (ftp://ftp.ncbi.nlm.nih. gov/pub/TraceDB/pan_troglodytes). There were a total of 101 Sanger-style trace read file sets available. Low quality bases were end-trimmed using a Phred value of 20, vector sequence was trimmed using the comprehensive NCBI ‘univ_vec_11-4-2014.fa’ file, and empty sequences and those less than 100 bases were discarded using the Lucy2 software package (Chou and Holmes 2001; Li and Chou 2004). The xml sequence information files were queried for run date information with minimum (beginning) and maximum (ending) run dates for experiments being parsed into an SQL table using a Python script written by this author. Only 84 of the 101 xml files contained information for run date.
After processing the trace reads, the average mean number of sequences per data set was 438,213 with a range of 46,251 to 496,267 sequences, and a median value of 470,609 sequences. Because each trace read file was on average extremely large, 25,000 test sequences were extracted at random from each data set and parsed into new FASTA format files using a Python script written by this author. The sequences were then queried against the hg19 version of the human genome using BLASTN v2.2.31 with the following parameters: evalue 0.1, word_size 11, outfmt 10, qseqid, qstart, qend, mismatch, gapopen, pident, nident, length, qlen, max_target_seqs 1, max_ hsps 1, dust no, soft_masking false, perc_identity 50, gapopen 3, gapextend 3, num_threads 10. Given the fact that previous versions of the BLASTN algorithm have had problems omitting query sequences, all data sets had the non-hitting sequences reblasted onto the human genome with the same parameters. In all cases, no further hits were obtained indicating that the BLASTN algorithm was not omitting query sequences as reported in previous versions (Tomkins 2015).
Resulting BLASTN output CSV format files were analyzed for a variety of basic statistical parameters and data visualization using a Python script written by this author. The CSV and FASTA files were also concatenated and imported into SQL tables for more detailed joins, views, and queries along with the run date information mentioned above. All Python parsing and analysis scripts, SQL table/ database generation Python scripts and SQL queries created by this author have been placed at github (https://github.com/jt-icr/chimp_trace_25k). Student T-tests for two-tailed, two-sample, unequal variance comparisons of datasets were done in Excel using the T.TEST function.
Results
Overall statistics and trends
At present, there are 101 DNA sequence datasets available to the public that were produced using Sanger style sequencing technology that yielded much longer read lengths than current next generation technologies which produce a greater amount of total bulk sequence of much shorter lengths (Henson, Tischler, and Ning 2012; Mardis 2008). The longer the read, the easier it is to computationally assemble into contiguous genomic regions called sequencing contigs. Therefore, all 101 of these datasets were downloaded and the sequences end-trimmed for poor quality bases and cloning vector contamination.
After sequence trace read processing to remove low quality bases, short reads (less than 100 bases), and vector contamination, the 101 multi-fasta files were analyzed for basic statistics. The minimum file size contained 46,251 sequences while the maximum was 496,267 sequences. Sequence length varied between 100 and 2012 bases with an average (mean) of 704 bases. Given that a total of 44,259,587 sequences were available from all 101 data sets, this represents a genome coverage of about 10.4 fold (after read processing) assuming a chimpanzee genome size of about 3 billion base pairs. The chimpanzee genome publication from 2005 utilized about half this amount of coverage as they reported an initial draft genome of about 5-fold coverage (Mikkelsen et al. 2005).
To ascertain the quality of each chimpanzee end-trimmed dataset, 25,000 DNA sequences were selected from each FASTA file at random and queried against the human genome (version hg19) using the most recent version of the BLASTN algorithm (version 2.2.31+). Liberal gap extension parameters were employed to allow for the longest possible alignments. Total number of sequences examined in this study was over 2.5 million.
Because previous versions of the BLASTN algorithm are proven to omit query sequences when blasting using large query datasets (Tomkins 2015), non-hitting sequences were re-blasted to verify that the algorithm was working correctly. In all cases, none of the reblasted sequences provided hits as was the case in previous releases of the algorithm that exhibited the bug. Clearly, the bug has been fixed, perhaps due in part to complaints to the developer team at NCBI by this author.
Overall, the basic statistics for the 101 data sets as a whole were as follows: The average alignment identify was 96.3% with an average length of 677 bases and 27 bases on average not aligning. When the non-aligning bases in each read are included, the average identity for the reads that hit on human was 92.6%. These results conflict with the initially reported alignable identity of 98.5% given in the 2005 chimpanzee genome publication.
Interestingly, data analyzed as a whole taken from this study do tend to more closely agree with leading primate evolutionist Todd Preuss who stated, “It is now clear that the genetic differences between humans and chimpanzees are far more extensive than previously thought; their genomes are not 98% or 99% identical” and “One consequence of the numerous duplications, insertions, and deletions, is that the total DNA sequence similarity between humans and chimpanzees is not 98% to 99%, but instead closer to 95% to 96%” (Preuss 2012). Preuss then cites three publications supporting this claim (Britten 2002; Varka and Nelson 2007; Wetterbom et al. 2006)—a list that does not include the 2005 chimpanzee genome paper.
It is noteworthy that the alignable DNA similarity of about 96% omits sequences that are too dissimilar to align onto human and thus inflates the actual overall genome similarity between chimpanzees and human. When including all non-alignable sequence, overall chimpanzee DNA sequence identity is only 90.8% for all 101 data sets sampled. Interestingly, this estimate of about 90% overall genome similarity is similar to a previous study by this author using the chimpanzee genome assembly as a substrate (Tomkins 2015). However, upon further analysis of the data in the current study, even this estimate is shown to be suspicious and likely inflated due to fundamental problems in the assembly of the chimpanzee genome as described below.
When the 101 data sets were plotted, it was clear that a major difference existed between them for overall DNA similarity—a trend which generally corresponded with the progression of the data sets by file name (fig. 1). It was also apparent that many data sets had overall DNA identities below 90%. Therefore, the data were divided into two different bins corresponding to below 90% overall identity or above 90% overall identity. Fifty-seven of the data sets had overall identities above 90% and 44 were below 90%. The basic statistics for each are shown in Table 1. When the two data sets were compared using overall identity as the test variable in a two-sample T-test, they were significantly different from each other (P < 0.0000001).
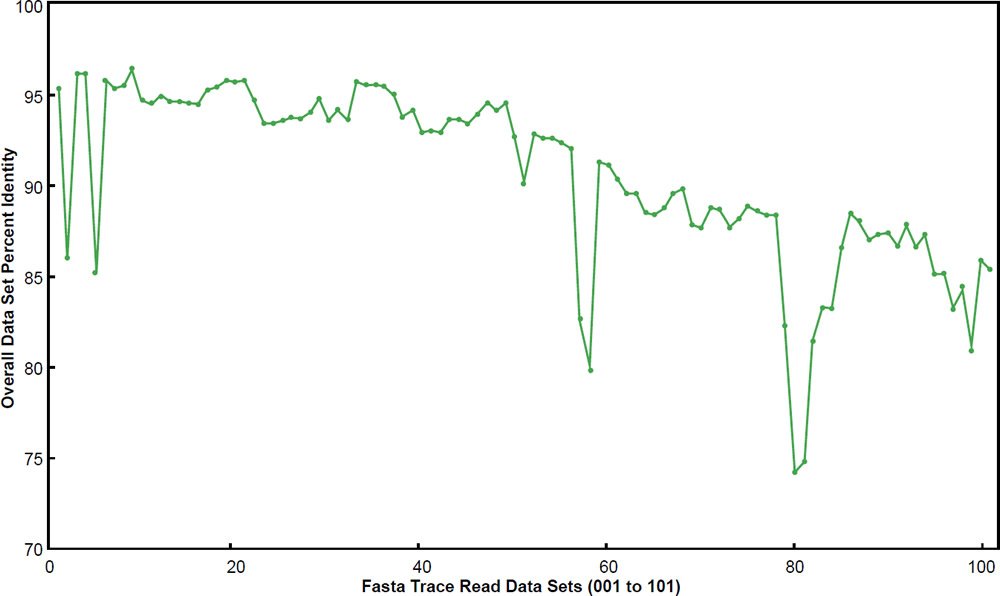
Fig. 1. Overall data set percent identity using BLASTN for the 101 chimpanzee trace read data sets compared to the human genome. Data sets are labeled 001 to 101.
| All Data Sets | High-Identity Sets | Low-Identity Sets | |
|---|---|---|---|
| Ave. Alignment identity | 96.3 | 96.7 | 95.8 |
| Ave. Query Seq. identity | 92.2 | 94.3 | 89.5 |
| Ave. Hit Frequency | 98.1 | 99.7 | 96.1 |
| Overall data set identity | 90.6 | 94.1 | 85.6 |
Human-Chimp DNA Similarity and Run Date
To determine if the apparent trend in reduced sequence similarity was associated with year of sequencing, run date information was extracted from each of the corresponding XML sequence information files. However, run date information was only recorded in 86 of the 101 XML files (85%). Furthermore, the run date data in each of the XML files in which it was recorded contained a range of years. Therefore, the SQL data tables extracted from the XML files were set up to include both the beginning and ending run dates. When queries were run to return information based on the ending year of sequencing, there were eight years involved between 2000 and 2011 (table 2).
| Sequencing End Year | No. Data Sets | Ave Alignment Identity (Percent) | Ave Query Seq Identity (Percent) | Overall Data Set Identity (Percent) | Percent Hits |
|---|---|---|---|---|---|
| NULL | 15 | 97.0 | 96.0 | 95.3 | 99.0 |
| 2002 | 10 | 97.1 | 93.8 | 93.7 | 99.1 |
| 2003 | 33 | 96.3 | 93.8 | 93.2 | 98.6 |
| 2004 | 21 | 95.6 | 89.8 | 89.1 | 98.5 |
| 2005 | 2 | 95.0 | 84.1 | 74.4 | 88.0 |
| 2006 | 13 | 96.2 | 92.1 | 86.7 | 93.6 |
| 2007 | 5 | 96.1 | 90.9 | 85.5 | 93.8 |
| 2008 | 1 | 96.0 | 92.2 | 86.2 | 93.0 |
| 2011 | 1 | 96.3 | 92.0 | 85.4 | 92.0 |
| 2002–2004 | 64 | 96.2 | 92.5 | 91.9 | 98.7 |
| 2005–2011 | 22 | 96.1 | 91.1 | 85.2 | 93.1 |
An approximately 5-fold genome coverage would have been obtained through the range of data sets completed through 2004, which would largely correspond with the data represented in the 2005 chimpanzee genome paper and the first initial draft of the chimpanzee genome. Therefore, these data were compared with those completed after 2004. The first set of sequences contained an overall DNA sequence identity (including non-hitting sequences) of 91.9% compared to 85.2% for the sequences corresponding to those completed after the first rough draft of the chimpanzee genome. When the two data sets were compared in a two-sample T-test, they were found to be significantly different from each other (P < 0.0000001).
Interestingly, the data sets completed through 2004 contained an average of 98.7% of the sequences providing hits onto the human genome. However, the data sets with completion dates of 2005 or later, only had a hit rate of 93.1%. A difference of 5.6% which was also significantly different in a two-sample T-Test (P < 0.0000001).
For the data sets with run date information, the progression of numbers in file names generally correspond well with the completion date of the data sets. It is clear from these analyses that the initial data sets used for the chimpanzee genome initial rough draft have significantly higher levels of DNA similarity than those produced later in the project (table 2). These early data would not only inflate the level of DNA similarity for chimpanzee compared to human as initially reported in 2005, but also bias the assembly and make it more human-like than it should be—compounding the problems caused by using the human genome as an assembly scaffold.
Another interesting aspect of this study was for the data sets that lacked information for run date—an oddity and indicator of sloppiness in the process of Sanger Style DNA sequencing (table 2). The recording of run date information in an accompanying xml sequence info file is a key factor in the trouble shooting of past sequencing runs. The data sets that lacked run date information were generally more similar to human than all the others in regard to alignment characteristics and had overall hit levels onto human of 99%. The corresponding file names (containing numbers in the range of 12 to 48) suggested that these data sets contributed to the initial 5-fold chimpanzee genome assembly. Of all the data sets evaluated, these had the highest levels of indicators for human DNA contamination.
Evaluating Chimpanzee Sequences Not Hitting Onto Human
Clearly, the data sets produced later in the chimpanzee genome project in the post-2005 genome paper timeframe, had much lower levels of hit percentages onto human. One question that arises in light of these results is whether these non-hitting sequences are of chimpanzee origin—a question that is difficult to answer given the questionable nature of the chimpanzee genome as an accurate substrate onto which they could be tested. Nevertheless, the chimpanzee sequences that had no hits on the human genome were blasted against the chimpanzee genome. The results were surprising and suggestive of miss-assembly in the chimpanzee genome due to a human framework bias by which it was constructed combined with the distinct possibility of assembly integration of human DNA contamination.
A total of 47,803 DNA sequences from the 2.5 million sequences sampled from the 101 data sets tested did not hit on the human genome. I refer to these as non-hitters. Using the same liberal BLASTN extension parameters as were done with human, 29,880 of the non-hitters (62.5%) provided hits onto the most recent version of the chimpanzee genome assembly at the time of this study (PanTro4), albeit at highly reduced identities with shorter alignments— compared to chimpanzee sequences that aligned to human.
When blasting chimpanzee trace reads onto an allegedly accurate representation of the chimpanzee genome, one would expect alignment identities of 99.9 to 100%. However, the average alignment identity (excluding all non-hitting sequence), was only 85.2%. These results strongly suggest that the chimpanzee genome is miss-assembled and more human-like than it should be.
Summary
In regard to data sets that included run date information, two different sets of chimpanzee DNA sequences related to the Sanger-style data sets used to construct the chimpanzee genome exist. The sequences that were produced early on in the chimpanzee genome project that contributed to the initial five-fold coverage of the chimpanzee draft genome (Mikkelsen et al. 2005), are significantly more similar to human than those that were produced later in the project by a difference of about 5% overall data set sequence identity. Contributing to this difference is the additional fact that a 5.6% difference in the amount of sequences that hit onto the human genome also exist.
When not considering run date, but instead including all sequences, two bins of data were constructed: data sets with overall identities below 90% and those above 90%. In doing this, the difference in sequence identity between the two data sets widened to 7%. This is largely due to the fact that the sequences lacking run date information were the most highly similar to human out of all the data sets. Because these data sets all contained filename numbers between 13 and 48, it is safe to assume that they contributed to the initial rough draft of the chimpanzee genome in 2005, inflating its human-like characteristics accordingly.
It may be that greater precautions towards human DNA contamination were taken later in the project producing less contamination. If the data from these seemingly less contaminated sets are considered, the chimpanzee genome is no more than about 85% similar to human. If all the data sets taken together are considered, despite the apparent human DNA contamination, then the chimpanzee genome is no more than about 90% similar to human.
It is very probable that the current chimpanzee genome assembly suffers from two major problems that make it more human-like that it should be. First, chimpanzee DNA sequences from both Sanger-style sequencing and next generation sequencing technologies, have been assembled using the human genome as a reference framework (Mikkelsen et al. 2005; Prado-Martinez et al. 2013). In other words, the chimpanzee genome does not stand on its own merits using its own framework-based genomic resources (e.g. an accurate integrated physical-genetic map for chimpanzee) as I described in an earlier publication (Tomkins 2011). Second, given the fact that significant levels of human DNA exist in non-primate databases due to laboratory and worker contamination (Longo et al. 2011), the potential for human DNA in the pre-assembled chimpanzee sequencing reads is highly probable and could be tested for by simply comparing the chimpanzee-human BLASTN analyses of the different data sets one to another. The main questions would be, are there significant differences between data sets, and are there any obvious patterns for these differences? The answer to both questions is a resounding yes.
In determining this, 101 Sanger-style publically available trace read data sets were downloaded, providing the longest possible trace read data source, were end-trimmed for low quality bases, and purged of contaminating plasmid cloning vector sequence. Then, 25,000 sequences were selected at random from each data set and queried against the human genome using BLASTN v2.2.31 with liberal gap extension. Results from the BLASTN analysis indicated that two different groups of chimpanzee DNA sequences existed. Those that were produced early in the chimpanzee genome project that contributed to the initial chimpanzee genome publication were considerably more similar to human than those that were produced later in the project by a difference of about 5%. It may be that greater measures towards alleviating human DNA contamination were performed as the project progressed. Data from the seemingly less contaminated sets indicate that the chimpanzee genome is no more than about 85% identical to human.
Furthermore, when chimpanzee sequences that did not hit onto the human genome were blasted against the chimpanzee assembly, the average alignment identity was only 85% when 99.9 to 100% identity should have been the result if the chimpanzee genome was accurately assembled.
References
Bradnam, K. R., et al. 2013. “Assemblathon 2: Evaluating de novo Methods of Genome Assembly in Three Vertebrate Species.” Gigascience 2 (1): 10.
Britten, R. J. 2002. “Divergence Between Samples of Chimpanzee and Human DNA Sequences is 5%, Counting Indels.” Proceedings of the National Academy of Sciences USA 99 (21): 13633–13635.
Chou, H. H., and M. H. Holmes. 2001. “DNA Sequence Quality Trimming and Vector Removal.” Bioinformatics 17 (12): 1093–1104.
Henson, J., G. Tischler, and Z. Ning. 2012. “Next- Generation Sequencing and Large Genome Assemblies.” Pharmacogenomics 13 (8): 901–915.
Li, S., and H.-H. Chou. 2004. “LUCY2: An Interactive DNA Sequence Quality Trimming and Vector Removal Tool.” Bioinformatics 20 (16): 2865–2866.
Longo, M. S., M. J. O’Neill, and R. J. O’Neill. 2011. “Abundant Human DNA Contamination Identified in Non-Primate Genome Databases.” PLoS One 6 (2): e16410.
Mardis, E. R. 2008. “Next-Generation DNA Sequencing Methods.” Annual Review of Genomics and Human Genetics 9: 387–402.
Mikkelsen, T., et al. 2005. “Initial Sequence of the Chimpanzee Genome and Comparison With the Human Genome.” Nature 437 (7055): 69–87.
Narzisi, G., and B. Mishra. 2011. “Comparing de novo Genome Assembly: The Long and Short of It.” PLoS One 6 (4): e19175.
Noonan, J. P. 2010. “Neanderthal Genomics and the Evolution of Modern Humans.” Genome Research 20 (5): 547–553.
Prado-Martinez, J., et al. 2013. “Great Ape Genetic Diversity and Population History.” Nature 499 (7459): 471–475.
Preuss, T. M. 2012. “Human Brain Evolution: From Gene Discovery To Phenotype Discovery.” Proceedings of the National Academy of Sciences USA 109, Suppl 1: 10709–10716.
Skoglund, P., B. H. Northoff, M. V. Shunkov, A. P. Derevianko, S. Pääbo, J. Krause, and M. Jakobsson. 2014. “Separating Endogenous Ancient DNA From Modern Day Contamination in a Siberian Neandertal.” Proceedings of the National Academy of Sciences USA 111 (6): 2229–2234.
Thomas, B., and J. Tomkins. 2014. “How Reliable are Genomes From Ancient DNA?” Journal of Creation 28 (3): 92–98.
Tomkins, J. 2011. “How Genomes Are Sequenced and Why It Matters: Implications for Studies in Comparative Genomics of Humans and Chimpanzees.” Answers Research Journal 4: 81–88.
Tomkins, J. P. 2015. “Documented Anomaly in Recent Versions of the BLASTN Algorithm and a Complete Reanalysis of Chimpanzee and Human Genome Wide DNA Similarity Using Nucmer and LASTZ.” Answers Research Journal 8: 379–390.
Varka, A. and D. L. Nelson. 2007. “Genomic Comparisons of Humans and Chimpanzees.” Annual Review of Anthropology 36: 191–209.
Wetterbom, A., M. Sevov, L. Cavelier, and T. F. Bergström. 2006. “Comparative Genomic Analysis of Human and Chimpanzee Indicates a Key Role for Indels in Primate Evolution.” Journal of Molecular Evolution 63 (5): 682–690.







