The views expressed in this paper are those of the writer(s) and are not necessarily those of the ARJ Editor or Answers in Genesis.
Abstract
The Nef protein of lentiviruses is a multifunctional protein involved in viral replication, immune evasion, and modulation of lymphocyte activation. It is considered the most important virulence factor of primate and human lentiviruses. Although the nef gene is conserved among SIVs and HIVs, only HIV-1 is highly virulent in its natural host. During the course of HIV-1 infection, the functions of Nef are modulated according to host environment. The ability of the Nef protein to down-regulate MHC class I and CD4 has been found to increase since the 1980s among North American patients. Comparison of Nef proteins from SIVcpz and HIV-1 indicates that after the virus crossed hosts from chimpanzee to human, Nef lost some of its potency to down-regulate CD28. There was also a loss of the ability of Nef to down-regulate TCR-CD3 before HIV-1 entered mankind. Down-regulation of CD3 and CD28 seem to affect the balance of the virus-host relationship. Loss of these functions, along with optimization of Nef to enhance viral replication in the new host species, has been selectively favored, resulting in short-term adaptation. It is suggested that this is a primary reason for the deadly nature of HIV-1 and the tragic AIDS pandemic in the human population.
Introduction
The pathogenesis of human immunodeficiency viruses (HIVs) remains speculative in spite of decades of intensive research. Nonhuman primates infected with simian immunodeficiency viruses (SIVs) have been used as animal models. SIVs and HIVs are structurally similar and both belong to the genus of lentiviruses. Genetic and epidemiological evidences suggest that HIV-1 originated from zoonotic infection of a chimpanzee strain of SIV (SIVcpz), while HIV-2 originated from SIV infecting sooty mangabeys (SIVsm) (Hahn et al. 2000). In contrast to human infections, most SIV infections are asymptomatic in their natural hosts (Silvestri et al. 2007).
One accessory protein of HIV-1, Nef, is thought to play a major role in the development of AIDS (Landi et al. 2011). The protein is conserved in all lentiviruses. It is rather small, consisting of 200 to 260 amino acids (Genbank). However, through a dazzling array of protein-protein interactions, Nef is found to play multiple roles in HIV infection, ranging from immune evasion to enhancement of replication efficiency (Foster and Garcia 2008). Interestingly, the functions of Nef are modulated through natural selection during the course of infection (Carl et al. 2001). Cyclic evolution and selection of Nef is akin to that of the gp120 Env protein in the periodic switch of coreceptor tropism (explained in a previous paper, see Liu 2015). The present paper focuses on evolution of the nef gene within a host, across hosts, and across species. Some of the mutations in nef appear to be irreversible and transmitted across hosts, contributing to genetic entropy of the SIV-HIV lineages. In contrast to the influenza viruses where genetic entropy resulted in decreased virulence (Carter and Sanford 2012), mutational degeneration of the nef gene manifests as increased pathogenicity of HIV-1.
Major Functions of Nef
1. Immune evasion
During the initial phase of HIV infection, the virus gains the upper hand against the host, replicating rapidly in infected cells and building up high viral loads in the patient’s blood. This initial expansion phase is eventually controlled by the host’s immune responses, followed by a long-term standoff between the virus and the host—the period of latency. A branch of the immune system, cytotoxic T lymphocytes (CTLs), plays a major offensive role in this battle, attacking and killing infected cells. In order for the CTL to distinguish between infected and uninfected cells, the infected cell must present fragments of viral proteins (peptides) on its surface. The viral peptides are transported to the cell membrane by the MHC class I proteins. The Nef protein of HIV foils this process by interacting with the MHC protein, and diverts it to the lysosomes for degradation (see fig. 1 which is reproduced from figure 1 of Hurley and Bonifacino 2012 at http://www.nature.com/nsmb/journal/v19/n7/images/nsmb.2304-F1.jpg).
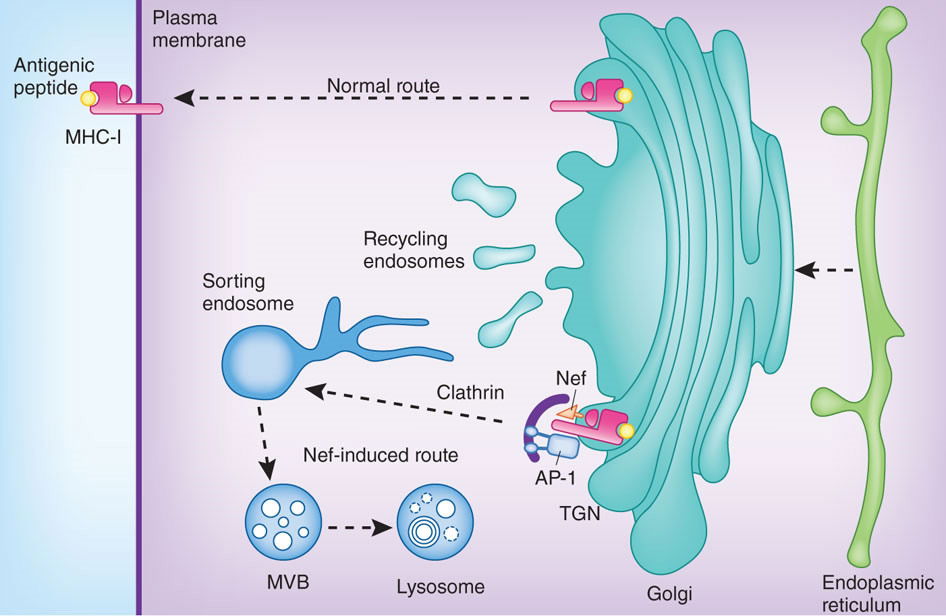
Figure 1. The Nef protein of HIV foils this process by interacting with the MHC protein, and diverts it to the lysosomes for degradation. Reproduced from figure 1 of Hurley and Bonifacino (2012).
2. Viral release
HIVs enter the cell through the CD4 receptor on the cell membrane. One function of Nef is to remove the CD4 receptor once the cell has been infected, so that when progeny viruses exit the cell they are not stuck on the membrane of the infected cell. Nef also removes coreceptors such as CXCR4 for the same purpose (Landi et al. 2011; Venzke et al. 2006).
3. Enhancement of viral infectivity
Experimental evidence points to Nef facilitating viral infection apart from its down-regulation of membrane receptors. The mechanisms are still speculative, including disruption of the cell’s actin cytoskeleton or interacting with unknown proteins (Foster and Garcia 2008).
4. Immune modulation
HIVs mainly infect helper T cells. Most of these cells are dormant, but viral replication demands active metabolism of the host cell. One of the functions of Nef is to activate the infected cell by interacting with host proteins involved in signal transduction (Markle, Mwimanzi, and Brockman 2013). However, activated T cells are short-lived, due to activation-induced cell death (AICD), which is a mechanism by which the body keeps immune responses under control. AICD is currently believed to be the main mechanism of T cell depletion in HIV infections, leading to AIDS (Paiardini and Müller-Trutwin 2013). It is not advantageous to the virus in the long run, because ACID leads to eventual death of the host. In order to effectively harness the infected cell for viral replication, the virus needs to strike a balance between host cell activation and activation-induced cell death. The Nef protein of HIV-2 and most SIVs is endowed with the ability to down-regulate the T cell receptor-CD3 complex (TCR-CD3). This prevents the T cell from being activated by external antigens binding the TCR. Besides the TCR, T cell activation also needs co-stimulation through the CD28 receptor. All primate lentiviruses down-regulate CD28 to modulate T cell activation.
Intrahost Evolution of Nef
Viral DNA preserved in latency
The viral enzyme for genome replication, the reverse transcriptase, is notoriously error-prone, leading to rapid diversification of viral genomic RNA. Even though strong selection preserves the infectivity of the progeny, lentiviruses depend on latent proviral DNA for long-term preservation. Some of the infected cells remain dormant for many years, with proviral DNA integrated in their chromosomes, providing a steady supply of non-degenerated viruses which can be transmitted into the next host (Salgado et al. 2010).
Powerful selection
A distinct advantage of viruses is their ability to preserve the integrity of their genomes through strong selection. Viruses typically produce a significant percentage of defective progeny, but their high reproduction rates afford strong selection. One indicator of purifying selection is the high ratio of synonymous versus nonsynonymous mutations in free viruses in the blood plasma of chronically infected hosts (Salgado et al. 2010). However, during the early phases, as the original founder virus diversifies, selection should favor mutants with enhanced activities that are beneficial at the moment. Indeed, positive selection on MHC down-regulation has been reported in the initial stages of HIV-1 infection (De La Cruz et al. 2014).
Lentiviruses are also capable of repairing mutations by selecting rare revertants. A 12-nucleotide deletion in the nef gene of an SIV was restored through duplications followed by multiple mutations (Whatmore et al. 1995). HIV-1 is also good at reverting substitution and frame-shift mutations in the nef gene (Watkins et al. 2013). Others reported compensatory mutations that partially restored the functionality of engineered nef mutants (Swigut et al. 2004).
Functional modulation of Nef via natural selection
The function of Nef is shaped by host environment. In the early stages of HIV infection, the CTL response effectively brings viral replication under control. Surviving viruses must be able to resist CTL, thus down-regulation of MHC class I is critical during this phase of infection. However, as the infection progresses from latency to AIDS, the CTL response eventually fails. Viral replication again goes unchecked. During the late stages, without immune selection, the ability of Nef to down-regulate MHC class I is gradually lost due to mutational degeneration. However, the ability of Nef to remove CD4 from infected cells, as well as its ability to enhance viral infectivity, still offers a competitive edge. Consequently, these activities experience positive selection and increase (Carl et al. 2001).
Opposite trends are seen in another group of patients. These individuals, called long-term nonprogressors, can mount a CTL response that effectively controls HIV replication over decades. In these patients, the ability of Nef to down-regulate MHC class I is maintained throughout the course of infection. However, the nef gene is forced to mutate constantly to escape CTL attacks. The cost of the escape mutations is reduced ability to down-regulate CD4 (Carl et al. 2001).
Interhost Evolution of Nef
Long-term evolution of HIV-1 Nef
Interhost long-term evolution of HIV has gained increasing attention in recent years (see review by Fraser et al. 2014). In spite of rapid diversification within hosts, virulence of HIV-1 is maintained through successive cycles of infection. As the chief virulence factor of the virus, Nef lies at the center of this dilemma. As mentioned above, immune escape mutations challenge the long-term preservation of Nef functions. A proven mechanism whereby the viral genome is spared from attenuating mutations is through proviral DNA in resting cells. Nevertheless, at least in long-term nonprogressors, even the DNA form of HIV-1 evolves slowly over time. One report found comparable levels of synonymous and nonsynonymous mutations in proviral DNA, indicating random drift (Salgado et al. 2010).
When nef sequences of HIV-1 from historic (1979–1989) and modern (2000–2013) eras were compared, significantly more diversification was observed in modern nef sequences, indicating ongoing evolution in the human population. Functional analysis indicated a decline in the efficacy of Nef to down-regulate MHC class I and CD4 molecules between the 1960s and the 1980s, followed by a restoration period between the 1980s and the modern time (Cotton et al. 2014).
From SIVcpz to HIV-1
HIV-1 is highly homologous to SIVcpz, a virus naturally infecting chimpanzees. Moreover, HIV-1 can infect chimpanzees, but not monkeys. Epidemiological data also support the notion that HIV-1 evolved from cross-species infection of SIVcpz (Hahn et al. 2000). Chimpanzees infected by HIV-1 are mostly asymptomatic. In one long-term study, only one out of 12 HIV-1 infected chimpanzees developed AIDS (Novembre et al. 1997). Compared with HIV-1, SIVcpz seems to be more pathogenic in chimpanzees. In one long-term observation in the wild, there was a 10–16 fold increase in age-corrected death hazard among infected chimpanzees compared to uninfected animals (the death risk in HIV-1 infected humans is 18–60, while that of HIV-2 is 2–5) (Keele et al. 2009). There is also evidence that SIVcpz affected the population dynamics of chimpanzees in the wild (Rudicell et al. 2010). These data suggest that after SIVcpz crossed species and evolved as HIV-1 in human hosts, it lost pathogenicity in chimpanzees. However, when different strains of HIV-1 were allowed to recombine in one chimpanzee, the virus regained its virulence and caused AIDS in the chimpanzee. Blood of the afflicted animal induced rapid and sustained loss of CD4+ cells when transfused into another chimpanzee (Novembre et al. 1997).
Apparently, HIV-1 gained virulence in humans but lost virulence in chimpanzees. A comparative analysis of the Nef protein revealed that the SIVcpz is more potent in down-regulating CD28 than HIV-1 is (Kirchhoff et al. 2004). Since CD28 is involved in inhibiting the activation of T cells, and activated T cells are more permissive to viral replication, natural selection would favor the loss of this Nef activity. Failure to suppress T cell activation and optimization of infectivity underlie the increased virulence of HIV-1 in humans.
Neither SIVcpz nor HIV-1 can down-regulate TCR-CD3 with their Nef protein, while HIV-2 and most SIVs possess this activity. It is widely held that SIVcpz evolved from SIVs of smaller primates on which chimpanzee prey (Bailes et al. 2003). So the ability to down-regulate TCR-CD3 was apparently lost during SIV evolution. According to Schindler et al (2006), it was lost in two SIV clades. The loss of Nef activities to down-regulate CD3 and CD28 both accelerated the rate of viral replication, thus was favored by natural selection. However, uncontrolled activation of the infected lymphocyte led to rapid demise of the cell, rapid depletion of CD4+ cells from the host, and eventual death of the host organism. By comparison, the low virulence of HIV-2 may have to do with these activities of Nef being retained in the SIVsm–HIV-2 lineage.
Conclusion
Endowed with high reproduction rates, lentiviruses can afford strong natural selection, which has helped the viruses to adapt to new hosts. Mechanisms of adaptation include optimizing the functions of Nef to evade host immunity and to maximize viral replication capacity. However, the near-sighted nature of evolution has caused Nef to lose its ability to keep lymphocyte activation under control, resulting in activation-induced death of the host cell and eventual death of the host organism. The ability of Nef to down-regulate TCR-CD3 and CD28 seems to be a design feature of this type of virus that has enabled a harmonious relationship between the virus and the host. Unfortunately this design feature, which previously made the virus benign, appears to have degenerated due to genetic entropy. Further studies of the long-term evolution of the ability of HIV-1 and HIV-2 to modulate CD28 or CD3 will help testing this hypothesis.
Retroviruses may have been created “good,” but degeneration of the Nef protein illustrates a mechanism by which pathogenicity evolved after the Fall.
References
Bailes, E., F. Gao, F. Bibollet-Ruche, V. Courgnaud, M. Peeters, P. A. Marx, B. H. Hahn, and P. M. Sharp. 2003. “Hybrid Origin of SIV in Chimpanzees.” Science 300 (5626): 1713.
Carl, S., T. C. Greenough, M. Krumbiegel, M. Greenberg, J. Skowronski, J. L. Sullivan, and F. Kirchhoff. 2001. “Modulation of Different Human Immunodeficiency Virus Type 1 Nef Functions During Progression to AIDS.” Journal of Virology 75 (8): 3657–65.
Carter, R. W., and J. C. Sanford. 2012. “A New Look at an Old Virus: Patterns of Mutation Accumulation in the Human H1N1 Influenza Virus Since 1918.” Theoretical Biology and Medical Modeling 9: 42.
Cotton, L. A., X. T. Kuang, A. Q. Le, J. M. Carlson, B. Chan, D. R. Chopera, C. J. Brumme et al. 2014. “Genotypic and Functional Impact of HIV-1 Adaptation to Its Host Population during the North American Epidemic.” PloS Genetics 10 (4): e1004295.
De La Cruz, J., T. Vollbrecht, P. Frohnen, H. L. Ng, E. S. Daar, O. O. Yang, and M. J. Lewis. 2014. “Ineffectual Targeting of HIV-1 Nef by Cytotoxic T Lymphocytes in Acute Infection Results in No Functional Impairment or Viremia Reduction.” Journal of Virology 88 (14): 7881–92.
Foster, J. L., and J. V. Garcia. 2008. “HIV-1 Nef: At the Crossroads.” Retrovirology 5: 84.
Fraser, C., K. Lythgoe, G. E. Leventhal, G. Shirreff, T. D. Hollingsworth, S. Alizon, and S. Bonhoeffer. 2014. “Virulence and Pathogenesis of HIV-1 Infection: An Evolutionary Perspective.” Science 343 (6177): 1243727.
Hahn, B. H., G. M. Shaw, K. M. De Cock, and P. M. Sharp. 2000. “AIDS as a Zoonosis: Scientific and Public Health Implications.” Science 287 (5453): 607–14.
Hurley, J. H., and J. S. Bonifacino. 2012. “Nef-arious Goings-on at the Golgi.” Nature Structural & Molecular Biology 19 (7): 661–62.
Keele, B. F., J. H. Jones, K. A. Terio, J. D. Estes, R. S. Rudicell, M. L. Wilson, Y. Li et al. 2009. “Increased Mortality and AIDS-like Immunopathology in Wild Chimpanzees Infected with SIVcpz.” Nature 460 (7254): 515–19.
Kirchhoff, F., M. Schindler, N. Bailer, G. H. Renkema, K. Saksela, V. Knoop, M. C. Müller-Trutwin et al. 2004. “Nef Proteins from Simian Immunodeficiency Virus-Infected Chimpanzees Interact with p21-Activated Kinase 2 and Modulate Cell Surface Expression of Various Human Receptors.” Journal of Virology 78 (13): 6864–74.
Landi, A., V. Iannucci, A. Van Nuffel, P. Meuwissen, and B. Verhasselt. 2011. “One Protein to Rule Them All: Modulation of Cell Surface Receptors and Molecules by HIV Nef.” Current HIV Research 9 (7): 496–504.
Liu, Y. 2015. “Cyclic Selection in HIV–1 Tropism: Microevolution That is Going Nowhere.” Answers Research Journal 8: 199–202. https://answersingenesis.org/biology/disease/cyclic-selection-hiv-1-tropism/.
Markle, T. J., P. Mwimanzi, and M. A. Brockman. 2013. “HIV-1 Nef and T-cell Activation: A History of Contradictions.” Future Virology 8 (4): 391–404.
Novembre, F. J., M. Saucier, D. C. Anderson, S. A. Klumpp, S. P. O’Neil, C. R. Brown II, C. E. Hart, P. C. Guenthner, R. B. Swenson, and H. M. McClure. 1997. “Development of AIDS in a Chimpanzee Infected with Human Immunodeficiency Virus Type 1.” Journal of Virology 71 (5): 4086–91.
Paiardini, M., and M. Müller-Trutwin. 2013. “HIV-Associated Chronic Immune Activation.” Immunology Review 254 (1): 78–101.
Rudicell, R. S., J. Holland Jones, E. E. Wroblewski, G. H. Learn, Y. Li, J. D. Robertson, E. Greengrass et al. 2010. “Impact of Simian Immunodeficiency Virus Infection on Chimpanzee Population Dynamics.” PLoS Pathogens 6 (9): e1001116.
Salgado, M., T. P. Brennan, K. A. O’Connell, J. R. Bailey, S. C. Ray, R. F. Siliciano, and J. N. Blankson. 2010. “Evolution of the HIV-1 nef Gene in HLA-B*57 Positive Elite Suppressors.” Retrovirology 7: 94.
Schindler, M., J. Munch, O. Kutsch, H. Li, M. L. Santiago, F. Bibollet-Ruche, M. C. Müller-Trutwin et al. 2006. “Nef-Mediated Suppression of T Cell Activation Was Lost in a Lentiviral Lineage That Gave Rise to HIV-1.” Cell 125 (6): 1055–67.
Silvestri, G., M. Paiardini, I. Pandrea, M. M. Lederman, and D. L. Sodora. 2007. “Understanding the Benign Nature of SIV Infection in Natural Hosts.” Journal of Clinical Investigation 117 (11): 3148–54.
Swigut, T., L. Alexander, J. Morgan, J. Lifson, K. G. Mansfield, S. Lang, R. P. Johnson, J. Skowronski, and R. Desrosiers. 2004. “Impact of Nef-Mediated Downregulation of Major Histocompatibility Complex Class I on Immune Response to Simian Immunodeficiency Virus.” Journal of Virology 78 (23): 13335–44.
Venzke, S., N. Michel, I. Allespach, O. T. Fackler, and O. T. Keppler. 2006. “Expression of Nef Fownregulates CXCR4, the Major Coreceptor of Human Immunodeficiency Virus, From the Surfaces of Target Cells and Thereby Enhances Resistance to Superinfection.” Journal of Virology 80 (22): 11141–52.
Watkins, R. L., W. Zou, P. W. Denton, J. F. Krisko, J. L. Foster, and J. V. Garcia. 2013. “In Vivo Analysis of Highly Conserved Nef Activities in HIV-1 Replication and Pathogenesis.” Retrovirology 10: 125.
Whatmore, A. M., N. Cook, G. A. Hall, S. Sharpe, E. W. Rud, and M. P. Cranage. 1995. “Repair and Evolution of Nef In Vivo Modulates Simian Immunodeficiency Virus Virulence.” Journal of Virology 69 (8): 5117–23.










{kind=link}